


▶Problem 1:
Generally, to fulfill the highly efficient anti-cancer drug delivery, a nanocarrier should be able to overcome the clearance effects from macrophages and penetrate into cancer cells in the tumor tissue. However, while neutral hydrophilic and negative-charged nanoparticles generally suffer much lower clearance effects than positive-charged and hydrophobic nanoparticles, it is usually the positive-charged nanoparticles that is favorable to cell internalization and endosomal escape. The contradiction in the favored properties at different delivery stages suggests that drug delivery systems of high efficacies cannot be based on conventional nanocarriers with invariable properties. Thus, a problem is demanding prompt solution: how to find a balance among these different requirements to fulfill the anti-cancer drug delivery?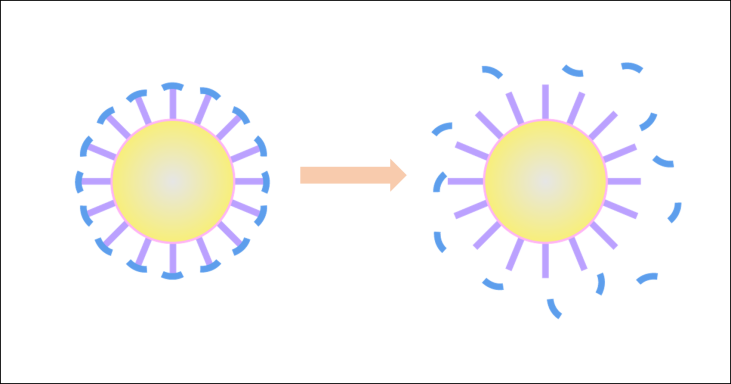
Scheme 1. Tumor microenvironment-activatable responsive drug carrier.
▶Solution 1:
An environmentally responsive drug carrier, triggered by the tumor microenvironment stimuli, could solve the problem. Here, we propose a nanocarrier with a zwitterionic surface to from a hydrating layer in blood circulation to resist the non-specific clearance by macrophage, while providing an enhanced half-life in blood circulation, however, the hydrating layer also prevent the cellular uptake. In the tumor micro-environment, distinguishing biological factors like over-expressed MMP-2 enzyme could remove the surface by clearing at the enzyme site. Thus, cell-penetrating blocks are exposed near the cancer cells and will fulfill the cellular uptake process.▶Problem 2:
As we discussed above, a drug carrier with environmentally responsive ability needs several functional segments and a hydrophobic block to drive the self-assembly. However, a single building block including all those parts can prevent conformational arrangement and correctly folding of chains during micelle formation.[6] Besides, a long and thus complicated peptide chain might also exhibit a random-coiled conformation after it is synthesized, which makes it much more difficult to assemble. How to achieve the various biological functions while ensuring the micelle formation?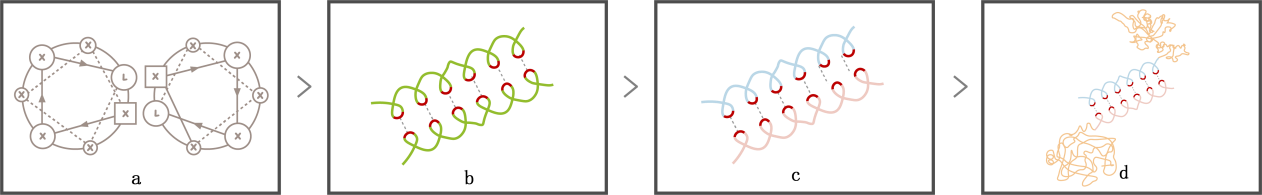
Scheme 2. leucine zipper coiled-coil. (a) Helical wheel diagram of leucine zipper; (b) Interaction between leucine zipper coiled-coil; (c) Interaction between LZR and LZE; (d) Connection between two polypeptide chains.
▶Solution 2:
Separating the functional block and the hydrophobic block into two chains guarantees the correct folding and makes it easier for self-assembly. Connection between two chains can be achieved by leucine zipper coiled coils, which is one of the largest and most conserved transcription factors and repressors family in eukaryotes. Its characteristic leucine repeat (Leu-X6-Leu-X6-Leu-X6-Leu) forms an ɑ helix , with Leucine locating at one side of the helix and offering a strong hydrophobic interaction to form protein-protein dimer [7].Moreover, By rationally adding arginine and glutamic to respective chains, the arginine-rich basic leucine zipper (LZR) and glutamic acid-rich leucine zipper (LZE) combine tightly to form a coiled-coil “LZR-LZE” complex via the hydrophobic and electrostatic interactions between leucine zipper coiled-coils. The strong interactions between two chains result in a high affinity with extremely low dissociation constant (Kd ≈ 10−15 M), and since the affinity between LZR motifs is much weaker,[8] the “LZR-LZE” complex is more favorable than “LZR-LZR” or “LZE-LZE” complex. With the LZR-LZE connection between two chains, the functional block can connect to the hydrophobic block to form a protein complex. Additionally, this modification brings us an extra merit:
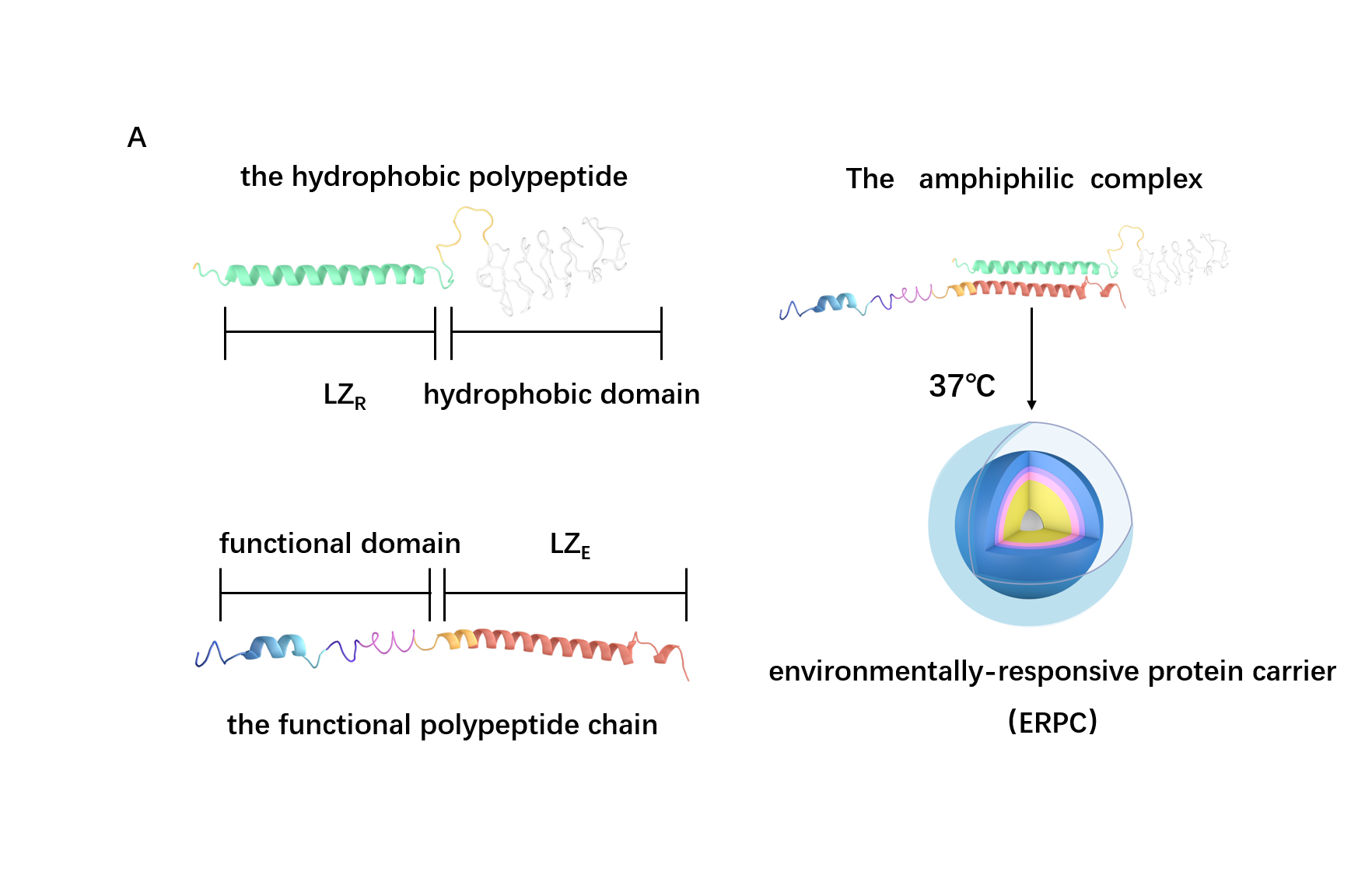
Scheme 3. The overall process of ERPC formation.
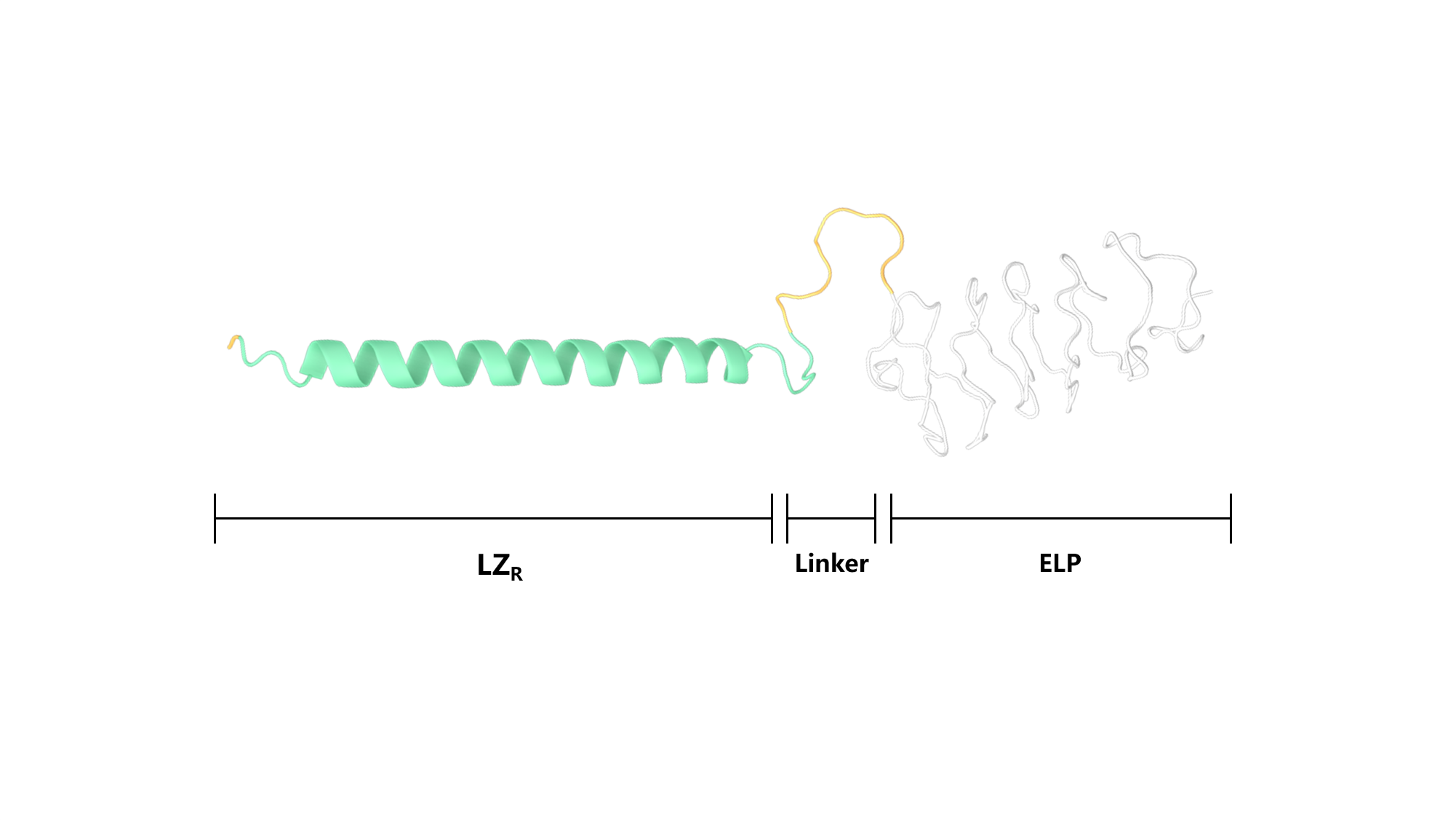
Scheme 4. Secondary structure of LZR-ELP.
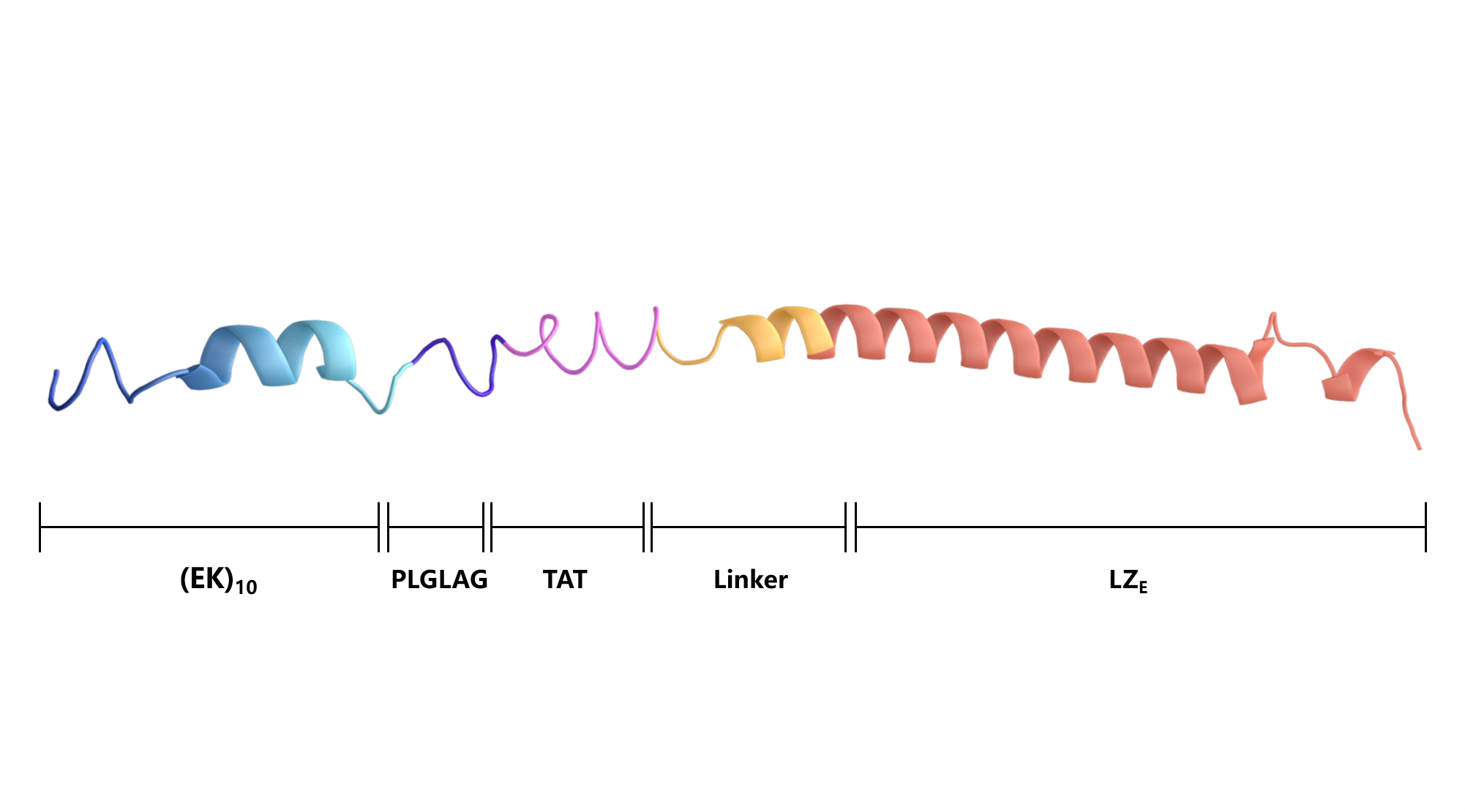
Scheme 5. Secondary structure of (EK)10-TAT-LZE.
Based on the structure of the two chains above, we can roughly predict the process of assembly into 2 steps: the dimerization of LZR-ELP and (EK)10-TAT-LZE and the assembly of ERPC.
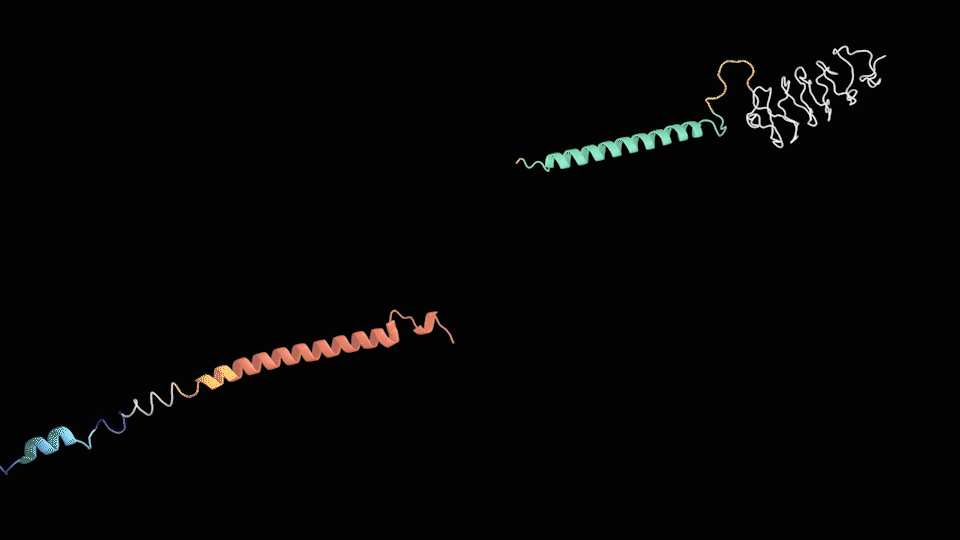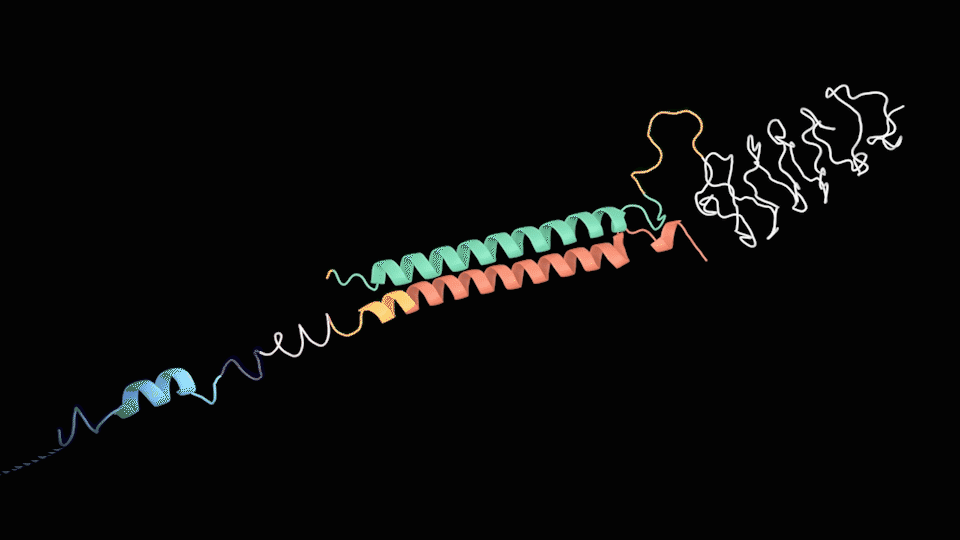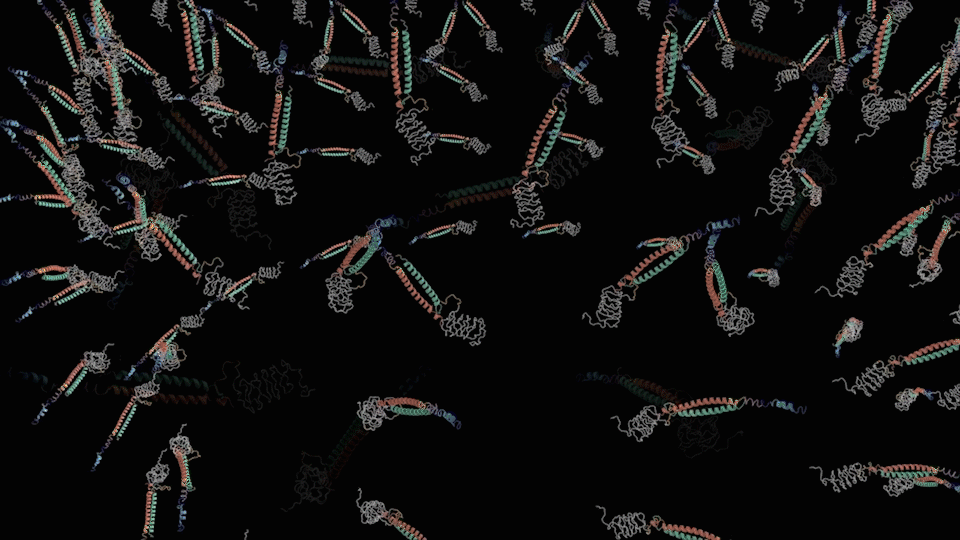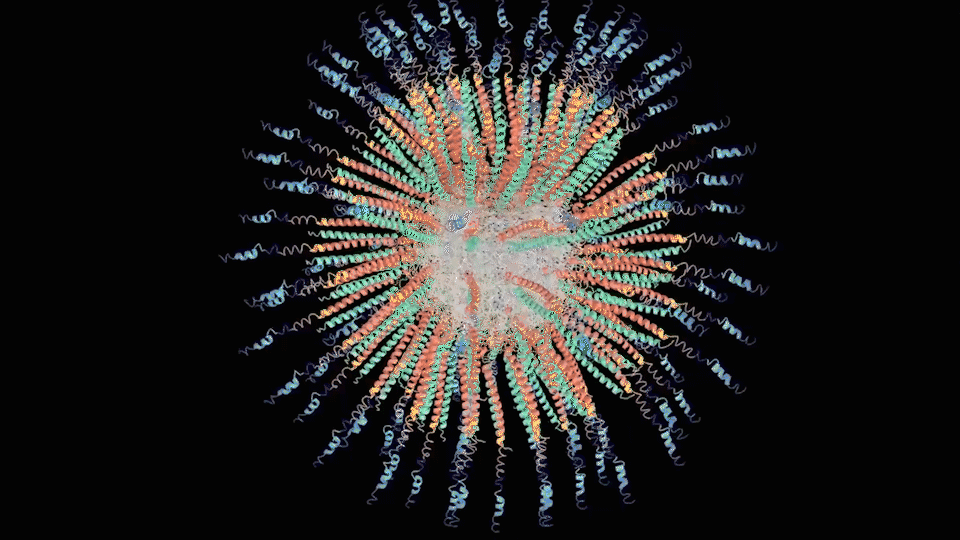
For the First step, the LZR-ELP and (EK)10-TAT-LZE dimerize via the hydrophobic and electrostatic interactions of leucine zipper coiled-coil (LZR/LZE) to form (EK)10-TAT-LZE/LZR-ELP amphiphilic complexes. During this process, the dispersed ELPs simplified the interaction in the hole system and makes it easier for the dimerization. For the second step, at room temperature, ELPs obtains a trend that gathering into a hydrophobic phase, leading to the amphiphilic complexes self-assemble into an environmentally responsive protein carrier (ERPC) nanoparticals.
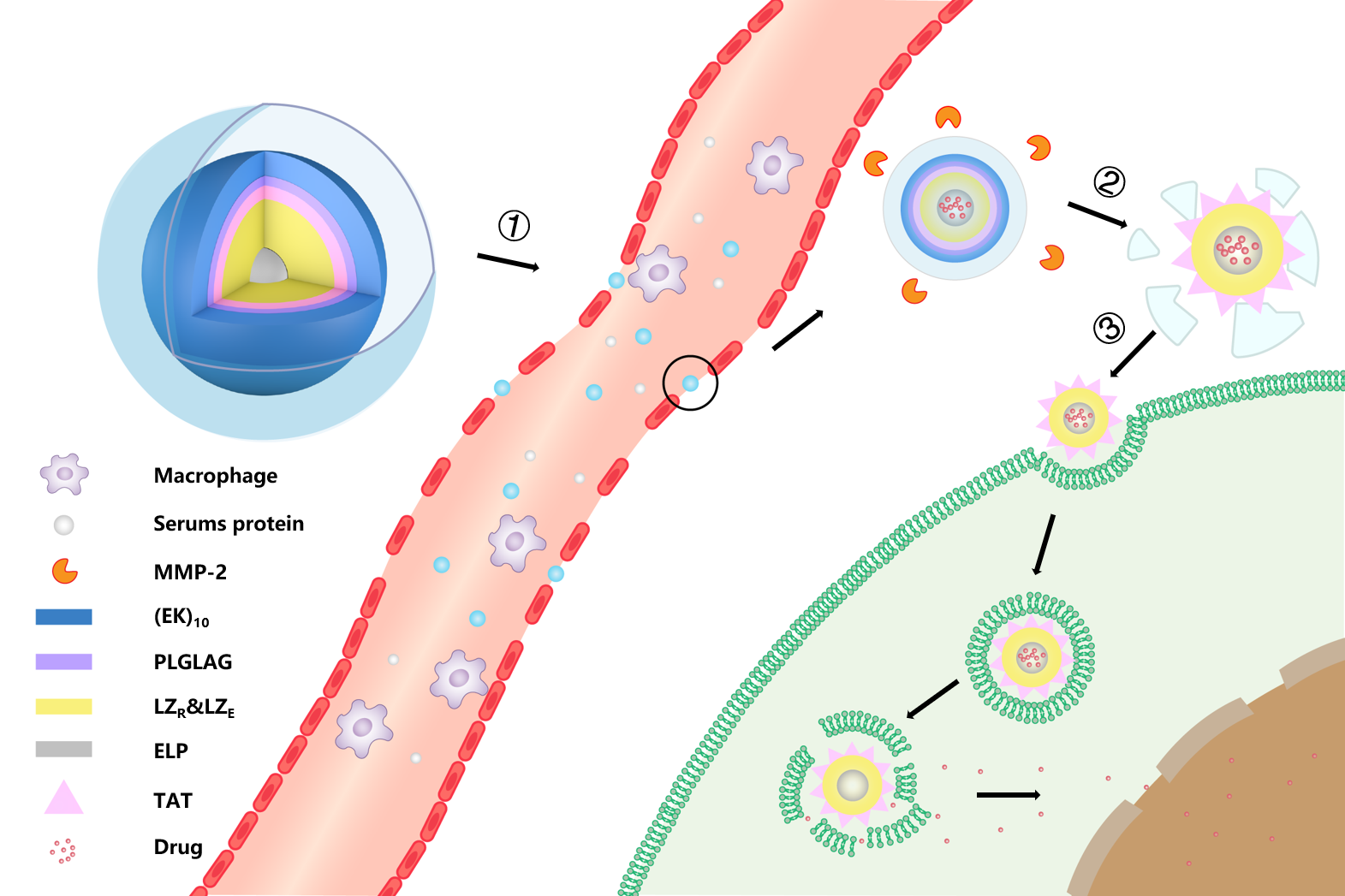
Scheme 6. biological function of EPRC.①Intravenous administration. ②MMP-2 enzyme clearance ③cellular uptake
Name | Sequence |
LZR-ELP | MALEIRAAALRRRNTALRTRVAELRQRVQRLRNEVSQYETRYGPL(G4S)5G[(VPGVG)2VPGFG(VPGVG)2]5 |
(EK)10-TAT-LZE | (EK)10PLGLAGYGRKKRRQRRRALMPVDGLEIEAAALEQENTALETEVAELEQEVQRLENIVSQYRTRYGPL |
Table 1. The sequence of LZR-ELP and (EK)10-TAT-LZE polypeptide.
ProCovar was used to predict the inter-residue contacts and secondary structure of LZR-ELP and (EK)10-TAT-LZE.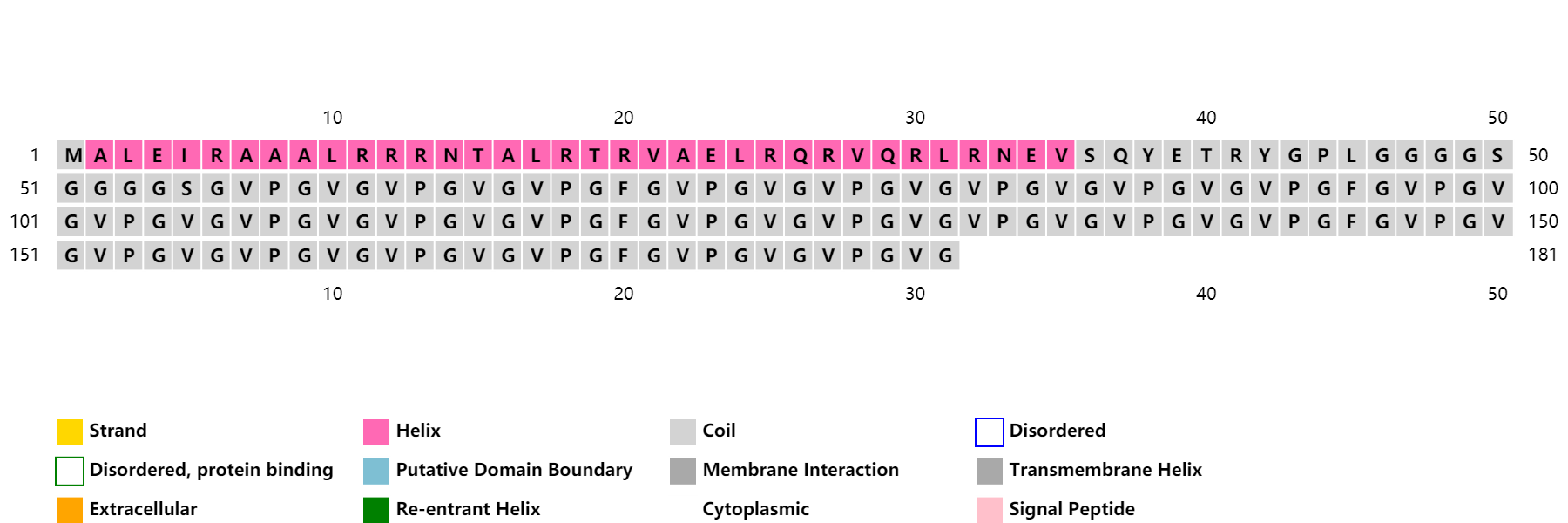
Scheme 7. Secondary structure prediction of LZR-ELP
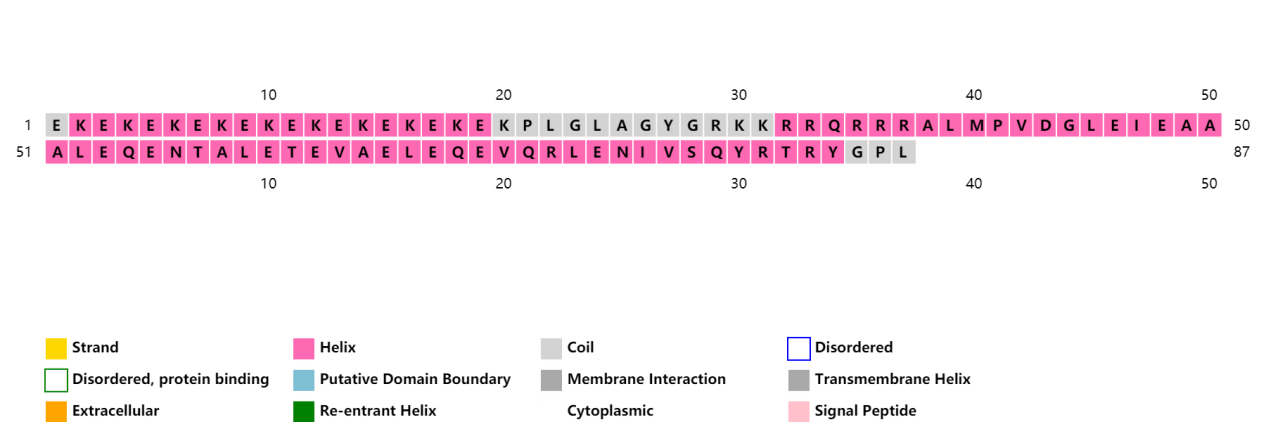
Scheme 8. Secondary structure prediction of (EK)10-TAT-LZE.[10-13]
We use SWISS-MODEL protein structure homology modelling to predict the interaction between LZR and LZE. Results were downloaded from SWISS-MODEL as a PDB file.
Scheme 9. Interaction between ZR and ZE[14-18]
Tertiary structure of LZR-ELP and (EK)10-TAT-LZE were predict using DEMO, a pipeline for constructing multidomain protein structures by docking-based domain assembly simulations. Results were downloaded from Zhang’s Lab as PDB files.
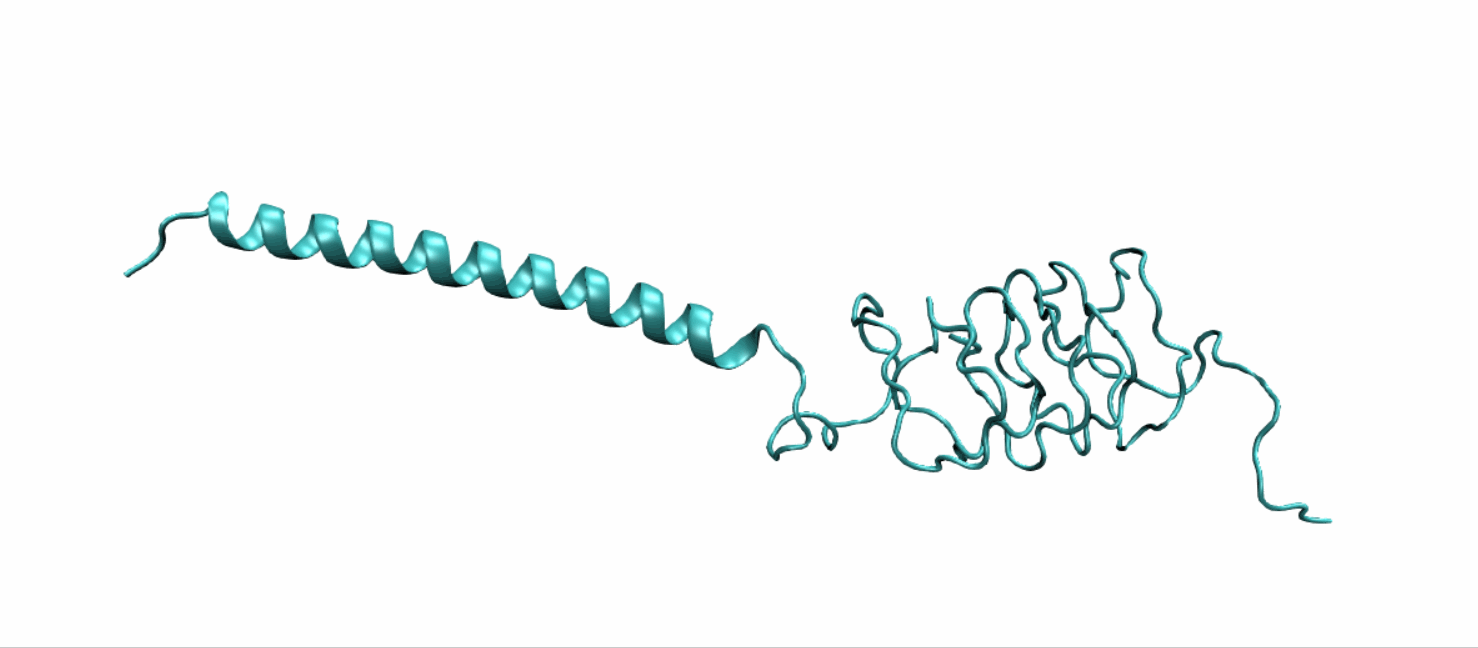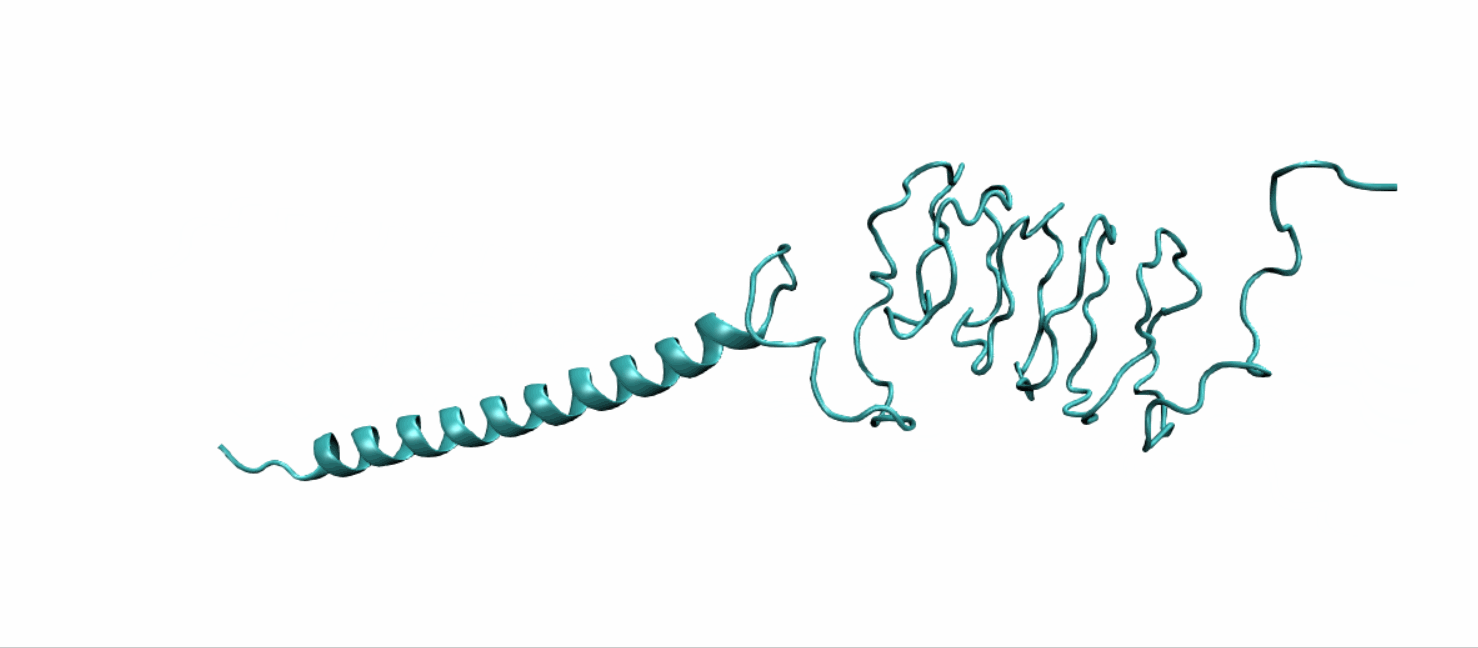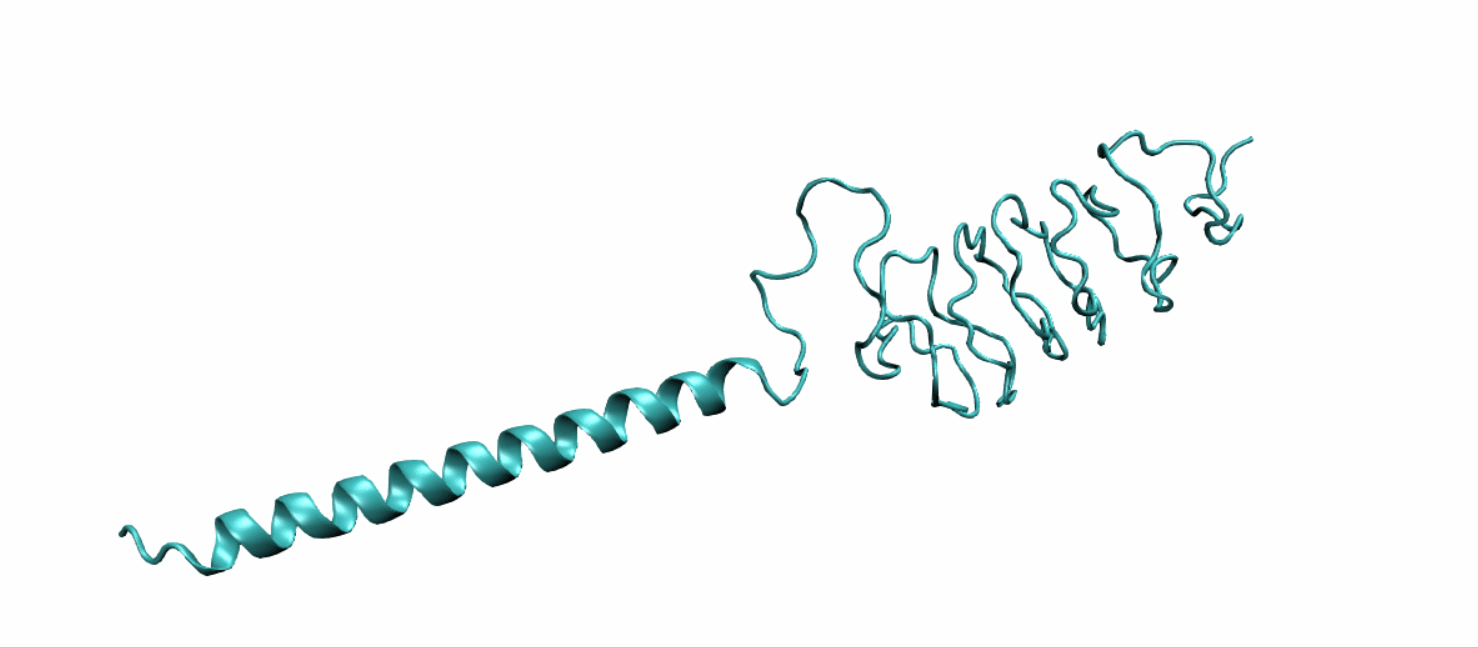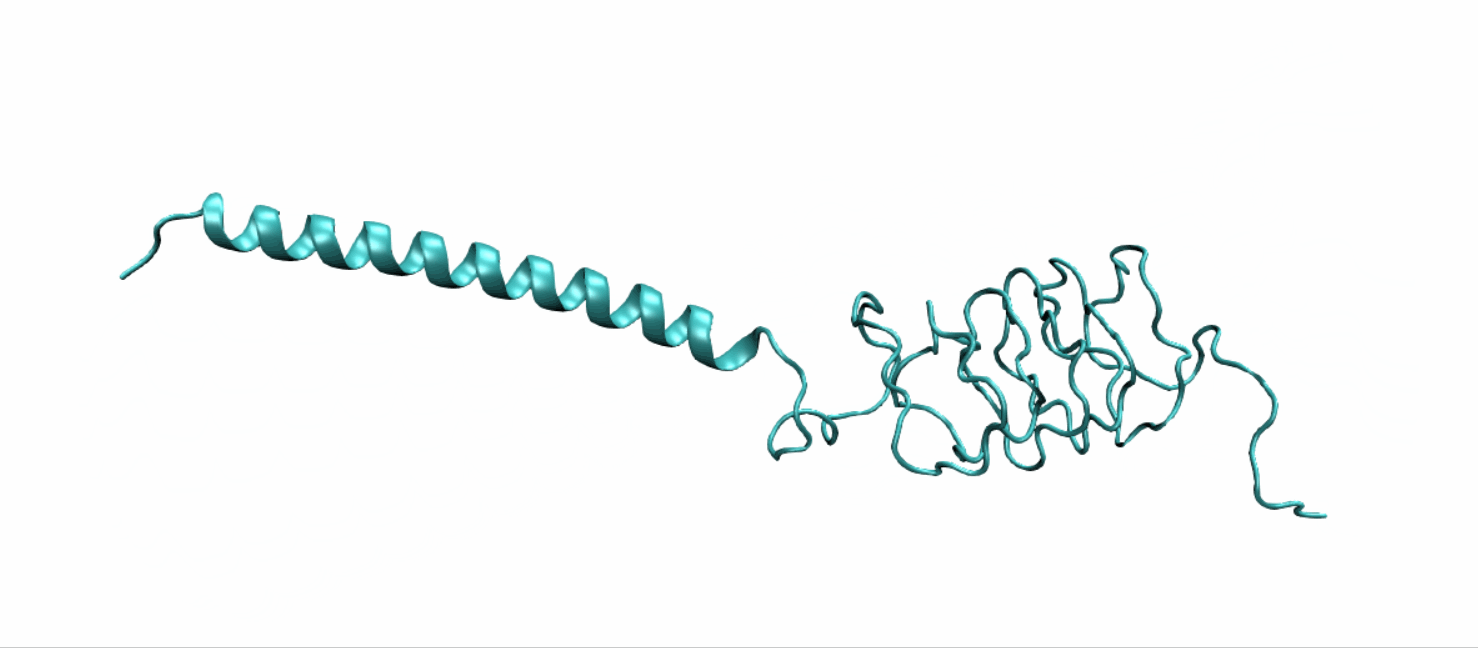
Scheme 10. Tertiary structure of LZR-ELP

Scheme 11. Tertiary structure of (EK)10-TAT-LZE[19]
The possibilities of residues in LZR-ELP and (EK)10-TAT-LZE being in contact were predicted using a heat map. “Being in contact” means the minimum backbone non-hydrogen atom distance of two residues falling in the range[0,6Å]. Predicted contact map was shown below and higher probabilities are represented by darker color.
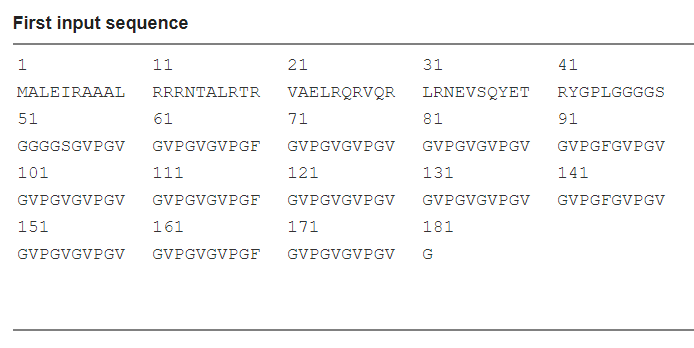
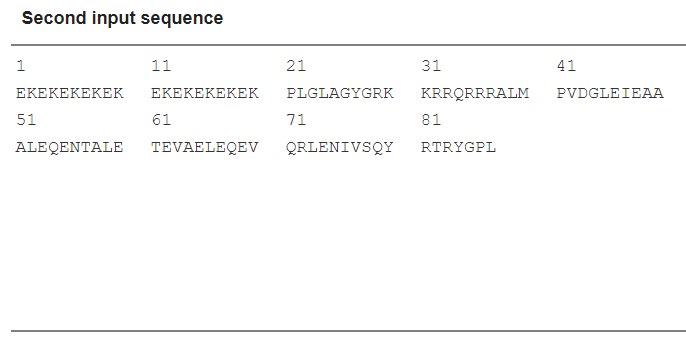
Scheme 12. Input sequence

Scheme 13. Predicted contact map of LZR-ELP and (EK)10-TAT-LZE membrane[20-23]
1. Expression and purification of LZR-ELP and (EK)10-TAT-ZE
2. Dimerization of LZR-ELP and (EK)10-TAT-ZE
3. Obtaining of ERPCs assemblies with fixed appearance.
4. Response to MMP2 enzyme
Goals for the future:
1. Loading and release of hydrophobic drug
2. Intracellular delivery of hydrophobic drug
3. In vivo anti-cancer drug delivery efficiency
AAFB~K$)I4~})K3Y.png)