We found the protein sequence of 4M, MG8 and Adhensin from NCBI. For the fusion protein, we added linker (GGGGS)3 to avoid the interaction between different component. The C-terminal of Adhensin is more active, so the C-terminal was reserved.


The gene encoding 4M, MG8 was codon optimized and commercially synthesized by BGI's Gene (Beijing, China), and then cloned into the NcoI and XhoI sites of pET28a vector. Here are the mappings.


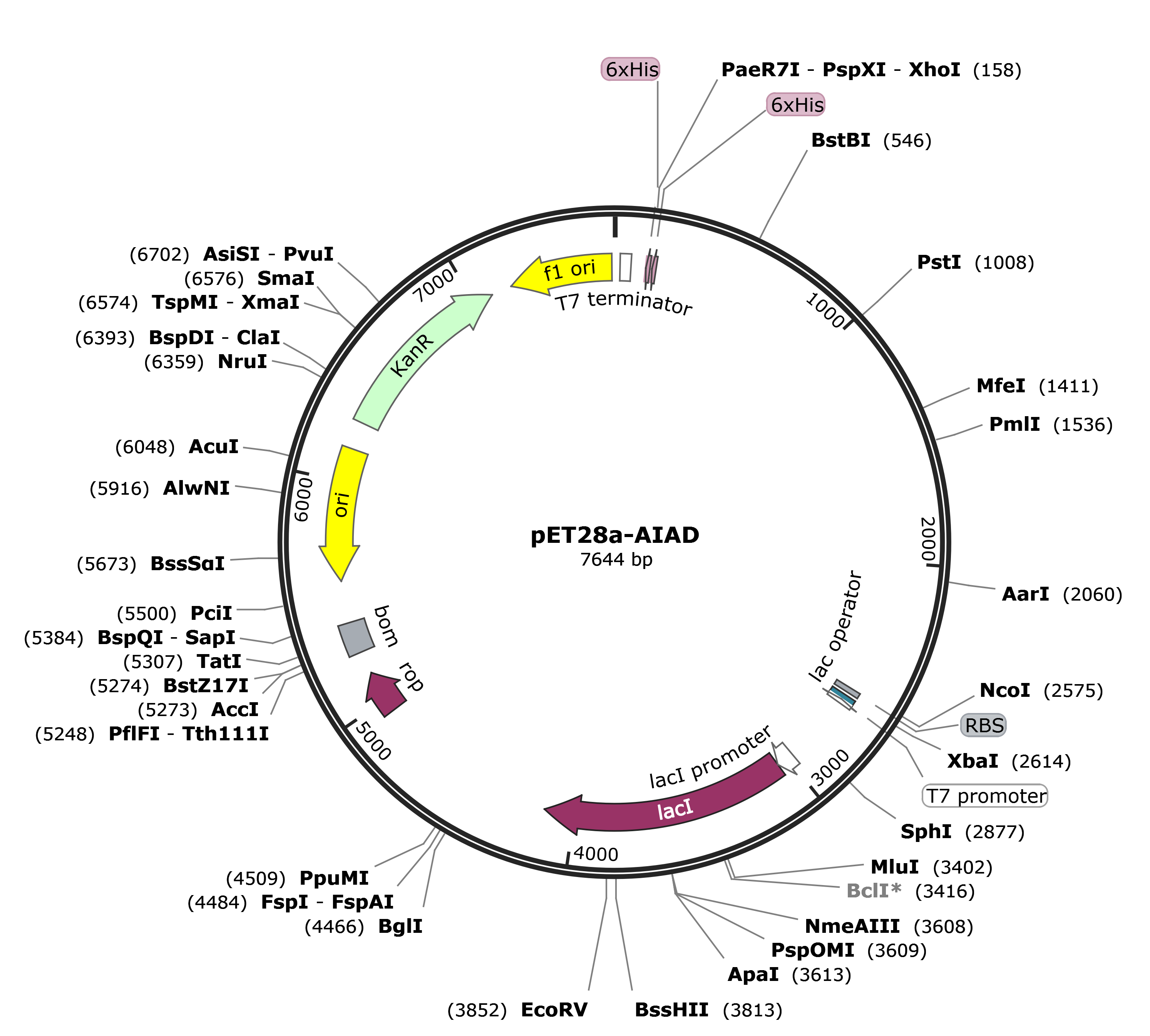
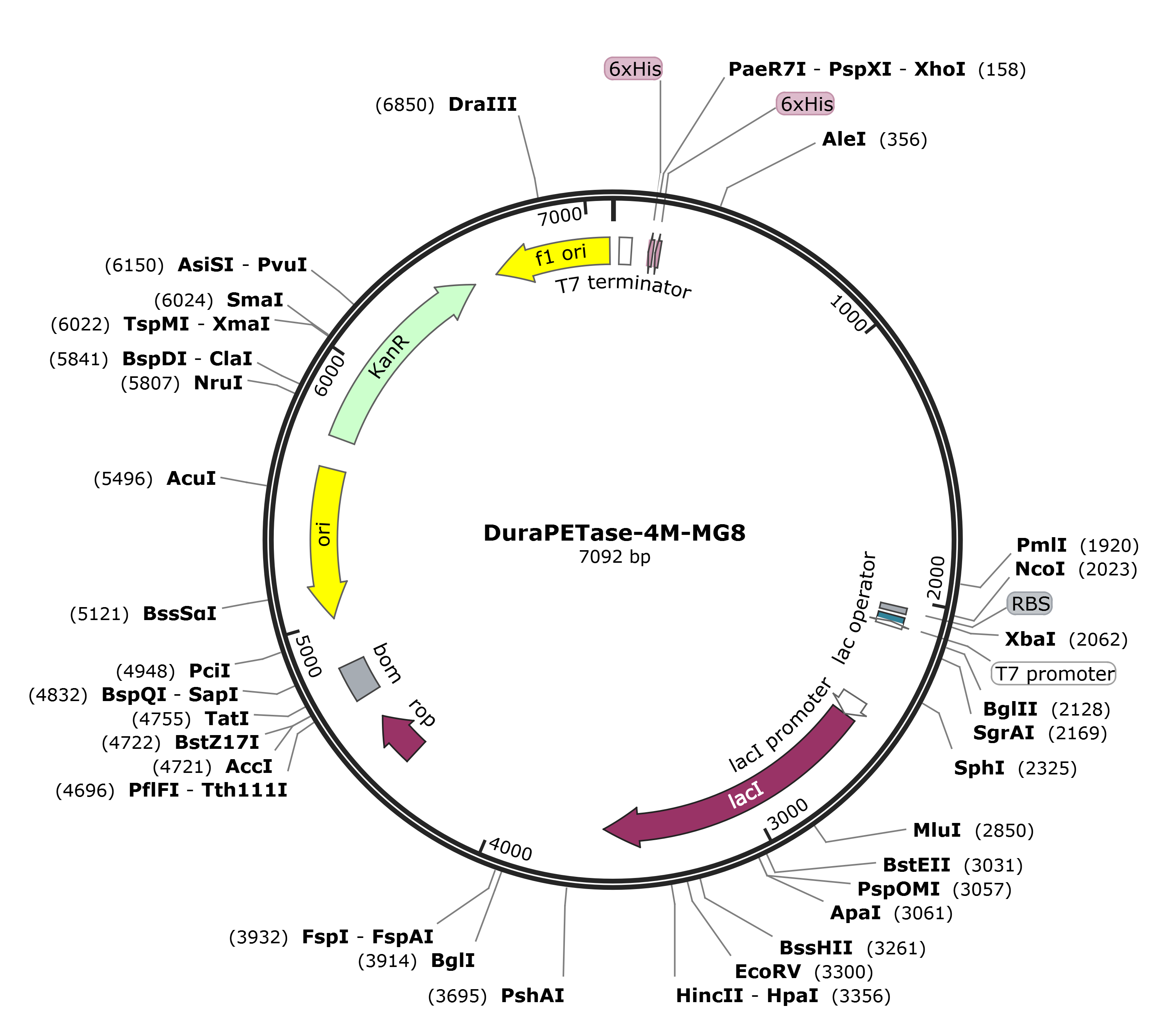
We received the plasmid dry powders and puncture bacteria of protein MG8-pET28a, 4M-pET28a, and 4M-MG8-pET28a produced by BGI's Gene.
Plasmid dry powder preparation before use:
The three plasmids (MG8-pET28a, 4M-pET28a, and 4M-MG8-pET28a) were centrifuged (12000 rpm, 1 min) and use 40μL of sterilized double-distilled water to dissolve. The dissolved plasmid concentration is 100 ng/μL.
The plasmids were transformed into competent states BL21(DE3).
Use and preservation of glycerol stock:
Dip the inoculation needle into glycerol stock and draw a line on the kanamycin resistant plate. The remaining glycerol stock were stored at -80℃.
Glycerol preserved three kinds of bacteria.
Pick up 3 normal growths of the colony to expand culture from each plate (MG8-DH5α, 4M-DH5α, and 4M-MG8-DH5α) in 10mL LB medium added 10μL kanamycin.
MG8, 4M, and 4M-MG8 were induced, adding 0.5mM IPTG induction (25℃, 220 rpm, 12h).
The plasmids (MG8, 4M, and 4M-MG8) were transformed into competent states BL21(DE3). Dip the inoculation needle into glycerol stock and draw a line on the kanamycin resistant plate.
Centrifuge (12,000 rpm, 30min, 4℃) and collect bacterial sediment.
Ultrasonic crushing, and then retain the supernatant and precipitate.
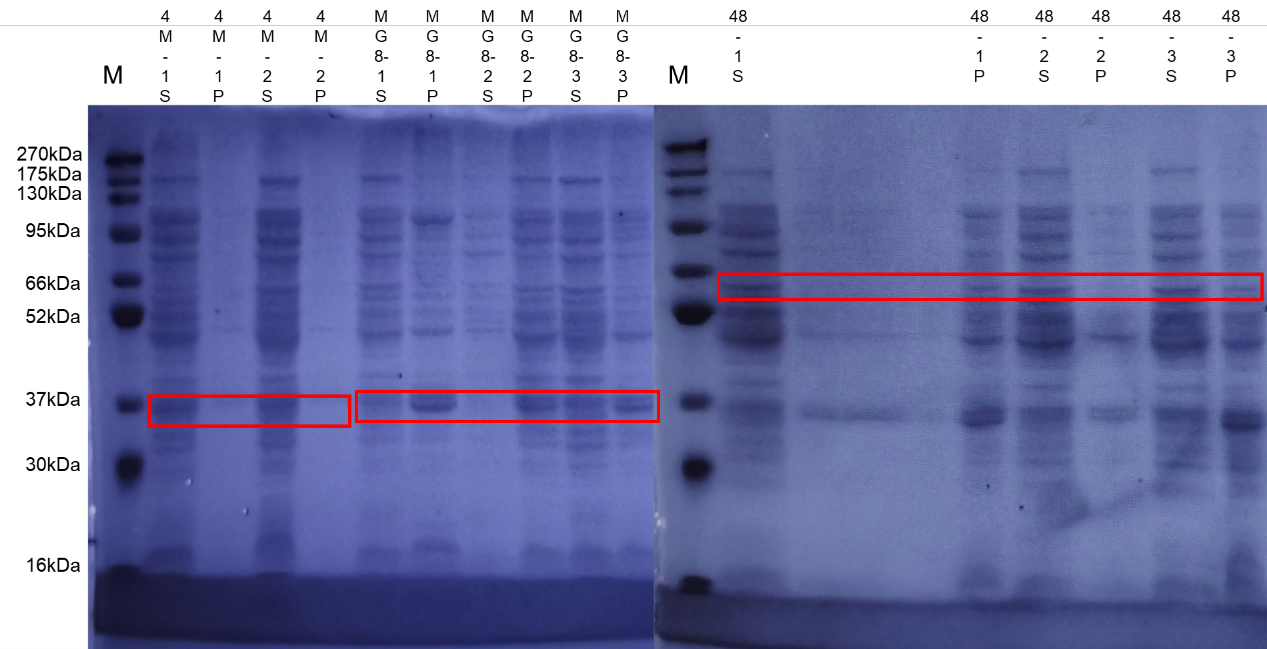
MG8, 4M, and 4M-MG8 induced, adding 0.5 volume of IPTG per thousand induction(37℃, 170 rpm, 14 h).
Centrifuge (12,000 rpm, 30min, 4℃) and collect bacterial sediment.
Ultrasonic crushing, and then retain the supernatant and precipitate.

Pick up 15μL of the preserved glycerol bacteria (4M and 4M-MG8) and cultured in 10 mL LB medium supplemented with 10μL kanamycin (37℃, 170rmp, 12h).
4M, and 4M-MG8 induced, adding 0.5 volume of IPTG per thousand induction (37℃, 170 rpm, 14 h).
Centrifuge (12,000 rpm, 30min, 4℃, four times) and collect bacterial sediment.
Ultrasonic crushing, and then retain the supernatant and precipitate.
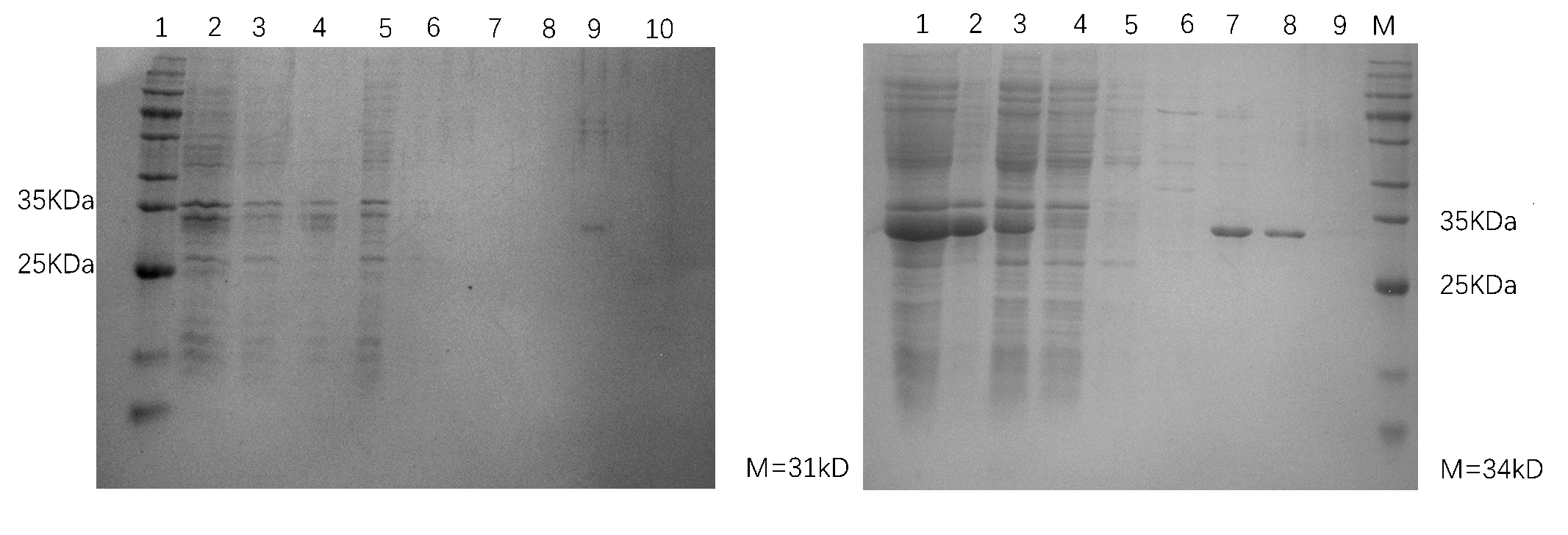
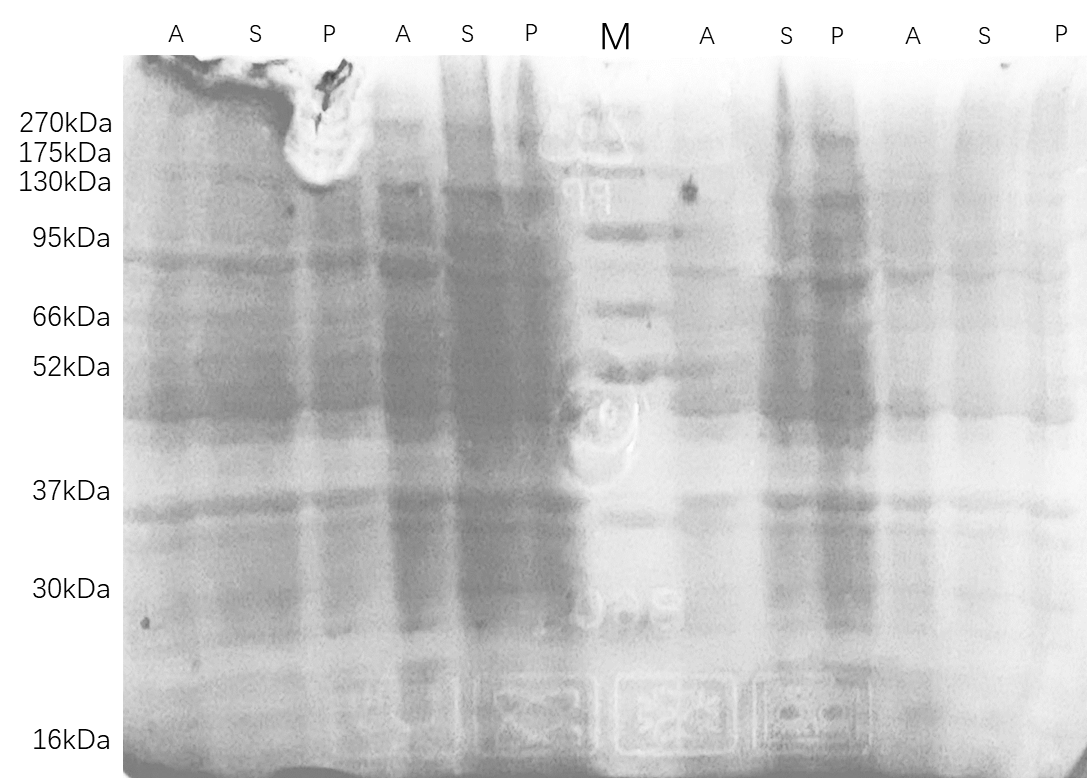
Supernatant and pellet/inclusion bodies were used for gel electrophoresis.
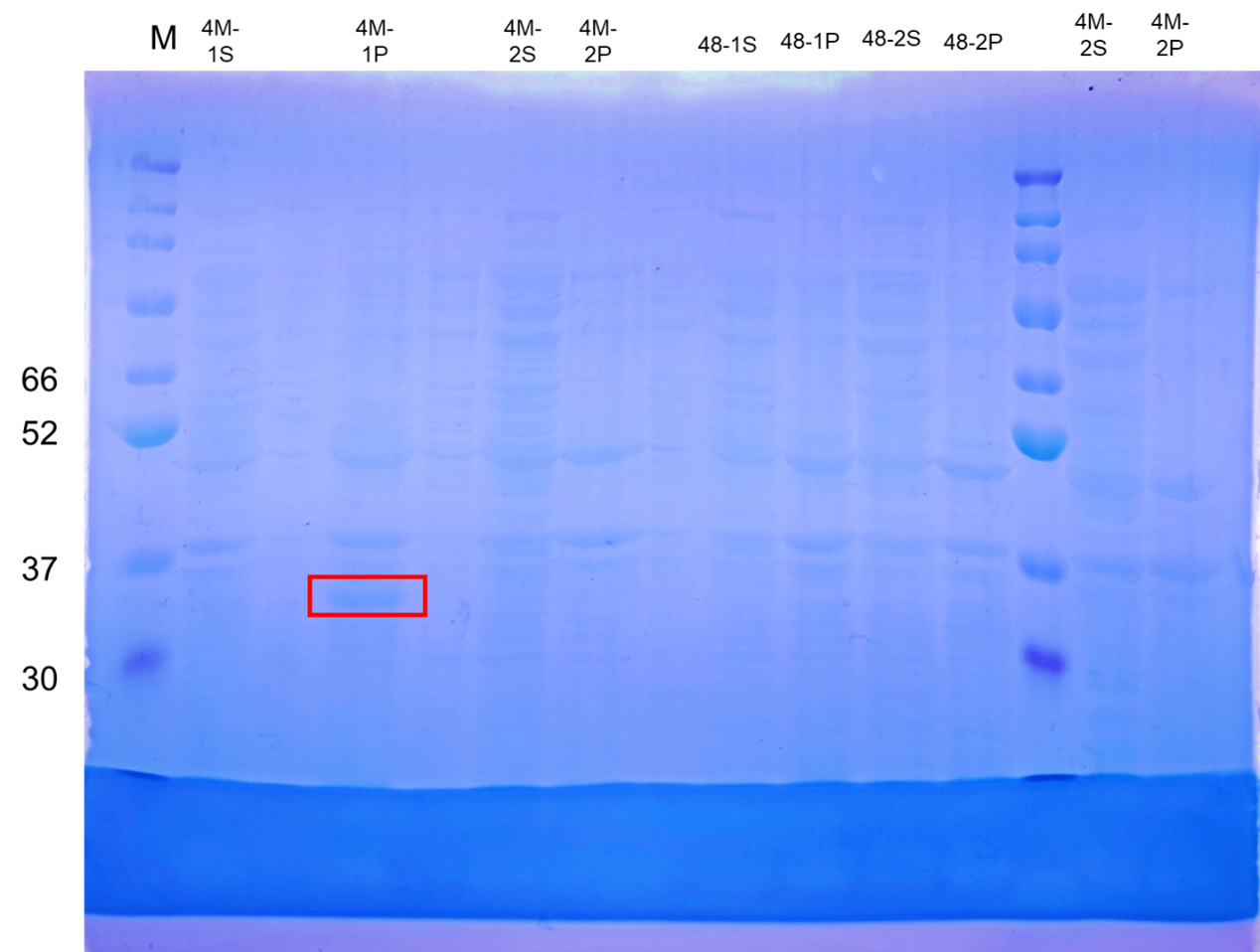
Plasmids MG8-pET28a, 4M-pET28a, and 4M-MG8-pET28a were extracted from MG8-pET28a-DH5α, 4M-pET28a-DH5α, and 4M-MG8-pET28a-DH5α.
Take 2mL of bacterial solution and centrifuge at 12000 rpm for 10min at room temperature to collect bacteria.
Discard the medium and add 500μL of solution I / RNase A mixture, vortex to make the cells completely suspended.
Transfer the solution to a new 2ml centrifuge tube and add 500μL. Gently invert and mix well, leave at room temperature for 2min.
Add 250μL of pre-cooled N3 Buffer and gently invert until a white flocculent precipitate is formed.
Centrifuge at 12000rpm for 2 min and remove the supernatant into a new centrifuge tube.
Adding 40 μL the magnetic beads. Before adding the magnetic beads make sure the beats are thoroughly mixed.
Incubate at room temperature for 10 minutes during which the mixture is mixed several times to prevent precipitation of magnetic beads.
Put the centrifuge into the magnetic rack magnetic suction until the solution is clarified and absorb the liquid in the tube.
Add 600μL 80% ethanol solution, mix thoroughly, put the centrifuge tube into the magnetic rack, magnetic suction until the solution is clarified absorb and discard the liquid in the tube.
Repeat last step, and try to suck up all the liquid in the tube.
Keep the centrifuge to on the magnetic rack with the tube cover open. Dry the magnetic beads for a few minutes until all the residual liquid evaporates. Observe the state of athletic beats and adjust the drying time appropriately according to the experimental environment. Dry until the surface of magnetic beads is dull. If drying is insufficient ethanol residue may affect downstream experiments.
Remove the centrifuge tube from the magnetic rack and add over 15μL of the eluate EL.
Mix thoroughly and incubate at room temperature for 5 minutes, during which mix several times to prevent precipitation of magnetic beads.
Place the centrifuge tube into the magnetic holder. Magnetic suction until the solution is clarified transfer the liquid out of the tube, and store for later activities.
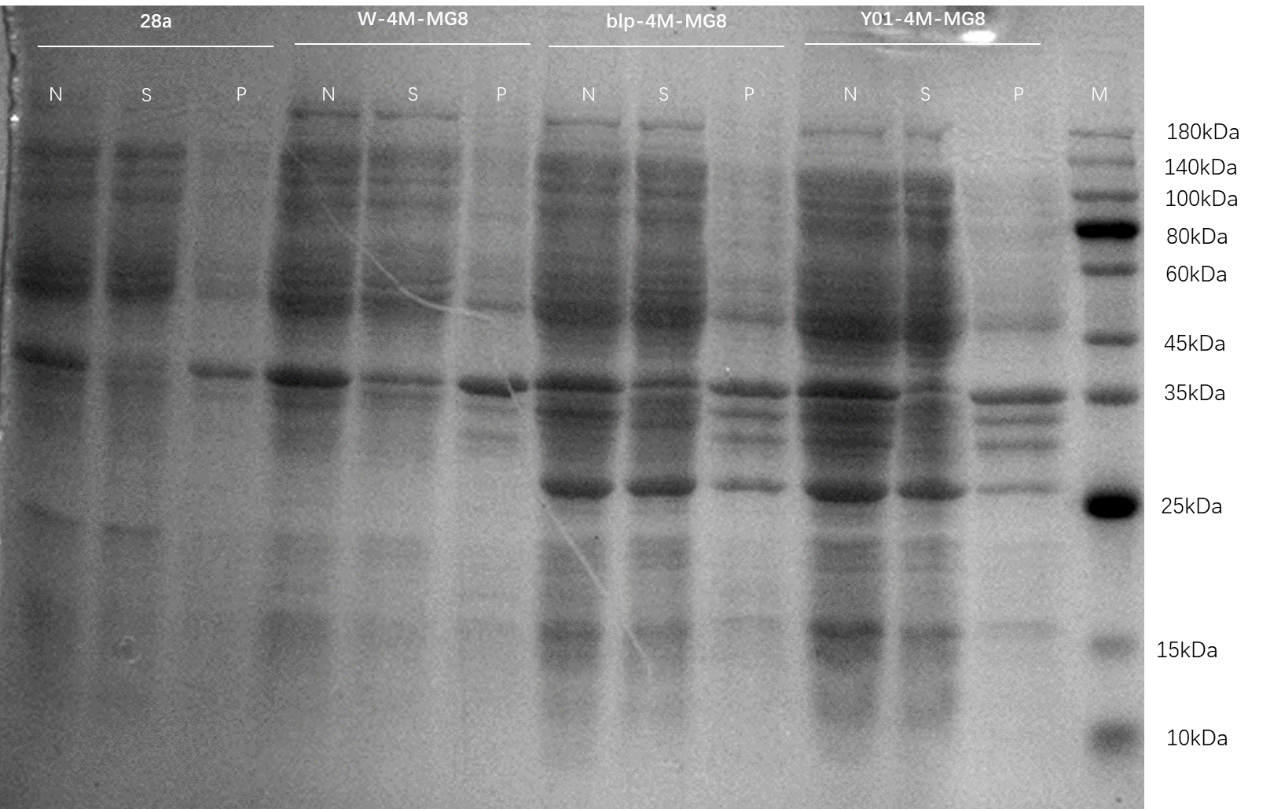
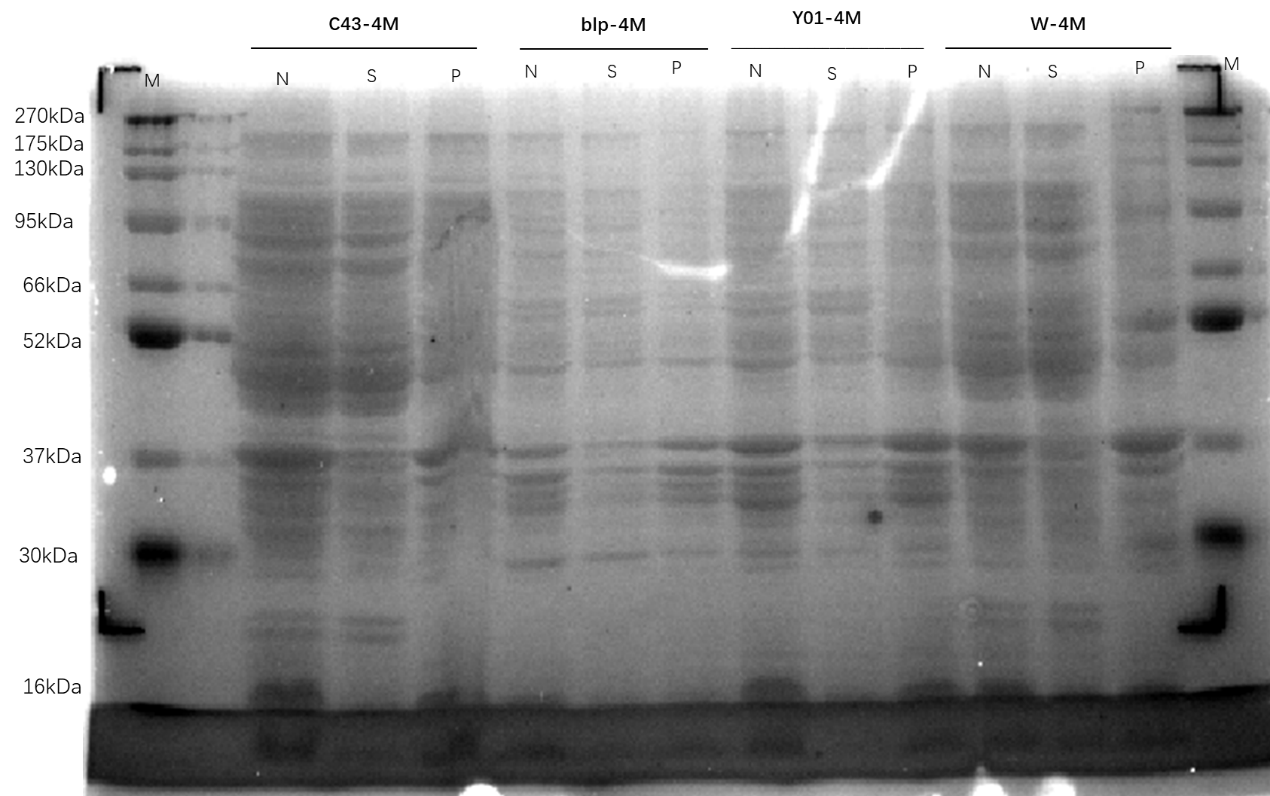
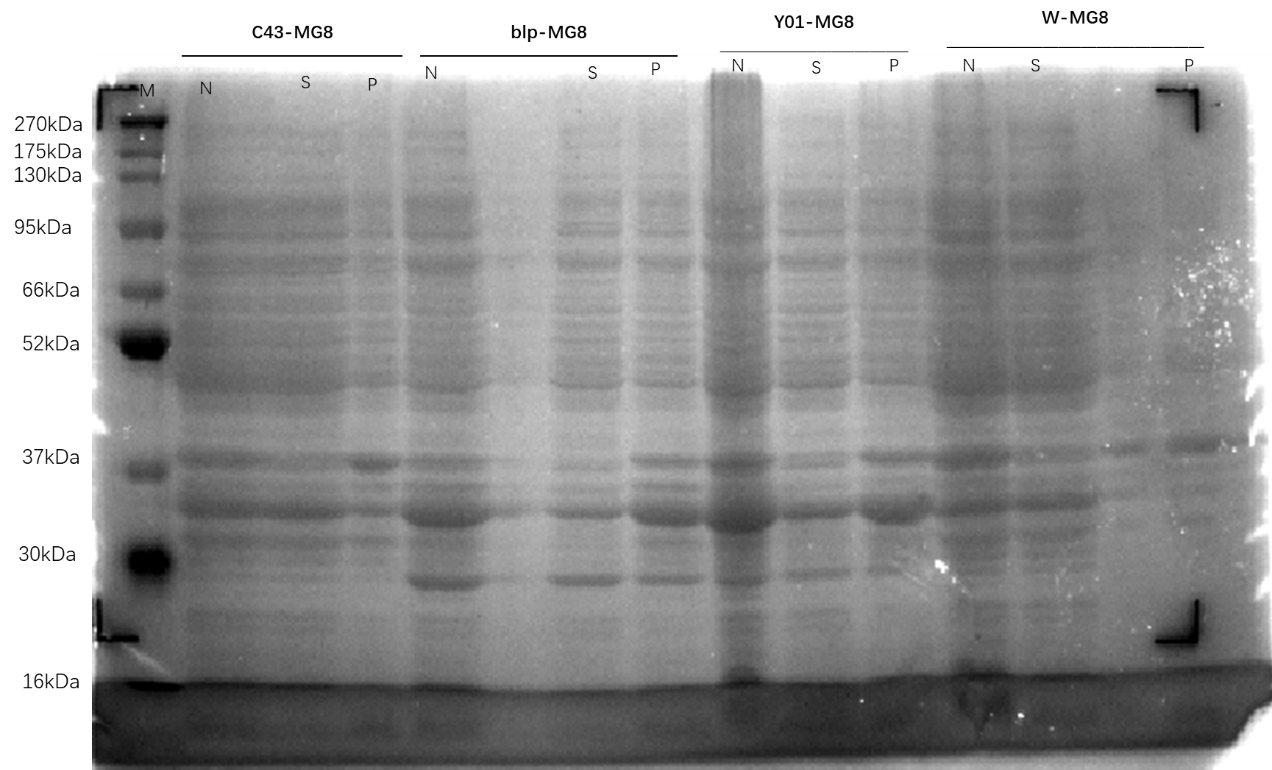
The plasmids MG8, 4M, and 4M-MG8 were transformed into competent states Y01, C43, W, BLP.
Pick up one normal growth of the colony to expand culture from each plate (MG8-DH5α, 4M-DH5α, and 4M-MG8-DH5α) in 10mL LB medium added 10μL kanamycin.
We received the plasmid dry powder and puncture bacteria of protein Adhesin produced by BGI's Gene.
Plasmid dry powder preparation before use:
The three plasmids Adhesin was centrifuged (12000 rpm, 1 min) and use 40 μL of sterilized double-distilled water to dissolve. The dissolved plasmid concentration is 100 ng/μL.
The plasmid was transformed into competent state BL21(DE3).
Use and preservation of glycerol stock:
Dip the inoculation needle into glycerol stock and draw a line on the kanamycin resistant plate. The remaining glycerol stock were stored at -80℃.
Adhesin-DH5α was used to extract plasmid of Adhesin later.
Transfer the bacteria in the test tube to 50mL LB medium ,added 50μL kanamycin.
MG8, 4M and 4M-MG8 induced, added 25μL IPTG. (25℃ 170rpm 14h)
Pick up one normal growth of the colony to expand culture in 10mL LB medium added 10μL kanamycin.
Centrifuge (12,000 rpm, 30min, 4℃) and collect bacterial sediment.
Ultrasonic crushing, and then retain the supernatant and precipitate.
Run SDS-PAGE to see the expression results.
Transfer the bacteria in the test tube to 100mL LB medium, added 100μLkanamycin.
Adhesin induced, added 25μL IPTG(25℃ 170rpm 14h).
After the ethanol flowed, add 10 times volume of water to wash.
Added 10 times column volume equilibrating solution, add crude enzyme solution to equilibrating column.
After 0.45μm filter membrane filtration, to hang the column, repeat three times, collect flow-through.
Added 12mL equilibrating solution, collect balance 5, add 10mL 10, 50, 100, 200mM imidazole to wash, and collect.
Add 10 times volume of water to wash.
20% ethanol to the column.
Centrifuge (12,000 rpm, 30min, 4℃) and collect bacterial sediment.
Ultrasonic crushing, and then retain the supernatant and precipitate.
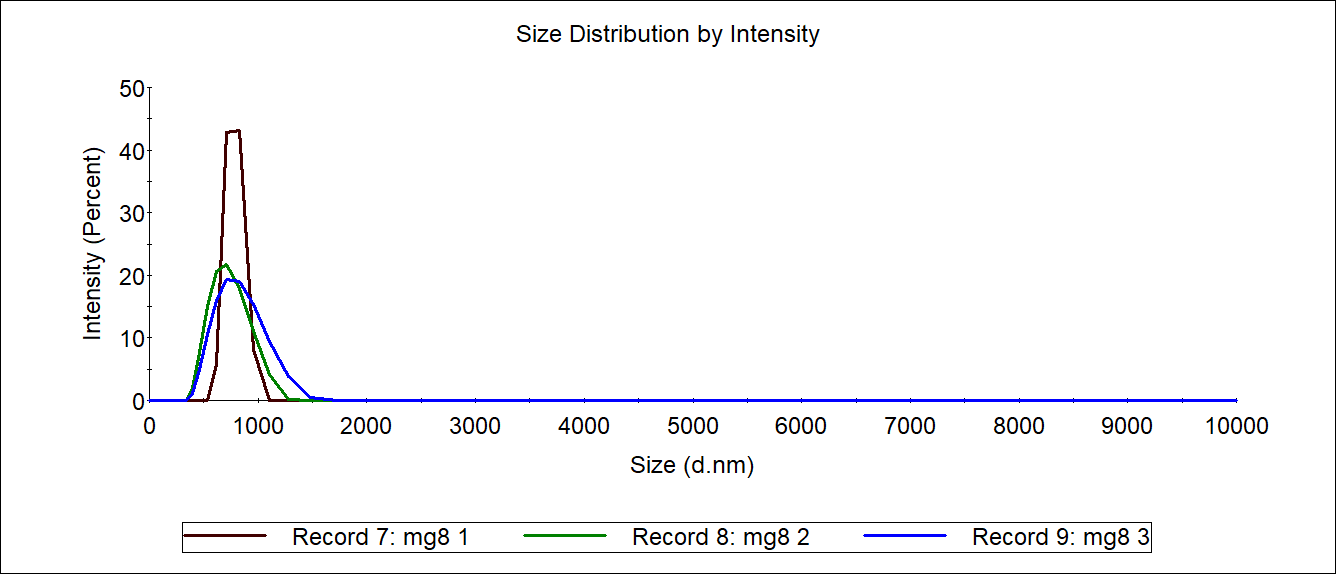
80μL Fe3O4 NPs (50mg/mL) were added in 2ml MES (50mM pH 5.5) buffer containing 5mg EDC and 4mg NHS and oscillated at 25℃ for 1 h at 150 rpm. Activated Fe3O4 NPs were magnetically recovered and washed three times with MES buffer. The activated carrier was re-suspended in 2mL 4M solution (0.1 mg/mL) and MG8 solution (0.1 mg/mL). Then they were incubated for 12 h at 4°C. The immobilized 4M (4M@Fe3O4) was magnetized and recovered and washed three times.
30μL 50mmol/L pNPA was added into 940μL Tris-HCl buffer (pH 8.0) as the substrate.
The enzyme was diluted in a gradient.
The original concentration of enzyme: 0.1mg/mL
The absorbance at 400nm was measured at 60℃ for 1min after decomposition.
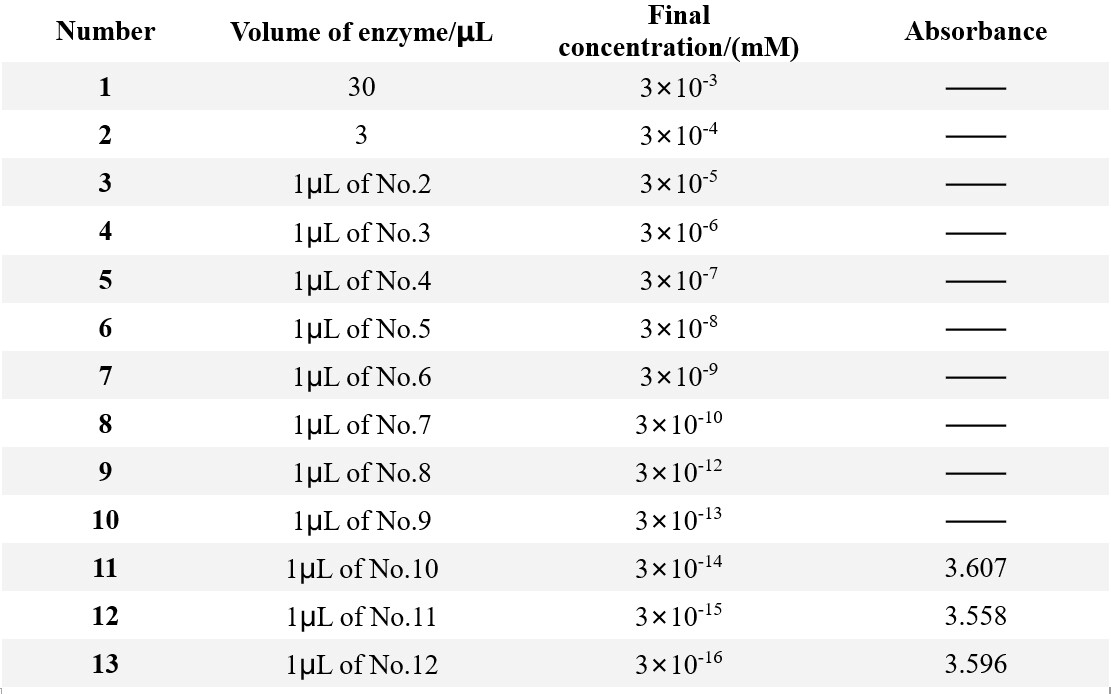
To quantify the amount of product p-NP, we measured the absorbance of different p-NP concentration.

After centrifugation, the clear supernatant was collected and subjected to affinity chromatography using a Ni2+-NTA column equilibrated with binding buffer (20mM Tris-HCl buffer, pH 8.0). Apply the supernatant to the column in binding buffer for three times, followed by washing of the column with washing buffer (10 mM, 50 mM, 100mM, 200mM, 500mM 1M imidazole). Flowthroughs from the column during collected.
1M imidazole solution: 17g imidazole, 250mL ddH2O
The remaining concentrations were obtained by gradient dilution:

Electrophoretic diagram of Adhesins
Adhesin was verified using 12% SDS–polyacrylamide gel electrophoresis (PAGE) followed by staining with Coomassie Blue R-250.
The remaining protein concentration was determined by BCA kit. A) Calculate the concentration of working liquid required for the experiment. When using microplate reader for detection, the required volume of working liquid for each test well is 200pl. Count the number of protein standard samples and egg samples to be tested (pay attention to the number of Wells required for parallel detection), and then multiply the total number by 200 to obtain the total volume of working liquid required (unit: μL).
B) BCA working solution is prepared according to the ratio of reagent (A) and reagent (b) volume ratio of 50:1, that is, 50 parts of reagent (A) and 1 part of reagent (b). The two reagents were thoroughly mixed, and the BCA working solution was stable within 24 hours at room temperature.

A) Add 20L of diluted protein standard solutions of different concentrations and protein samples to be tested into 96-well microplate in turn. Note If the protein sample concentration is too high, the protein sample should be diluted with the same solution as the standard protein dilution.
B) Add another 200! For BCA working solution, the enzyme-labeled Q plate can be used for functional shock for 30S, or the enzyme-labeled plate can be lightly flicked by hand to mix the solution. It was placed at 37℃ for 30min.
C) The absorbance value of 562nm was determined by microplate reader. The standard concentration of protein was taken as the abscess coordinate, and the absorbance value was taken as the ordinate coordinate. It was noted that the absorbance value of blank control should be subtracted from the absorbance value, and the standard curve was drawn to obtain the linear formula and R2 value.
D) According to the formula obtained in the previous step, calculate the concentration of the corresponding protein sample according to the absorbance value of the protein sample to be measured.
E) If the spectrophotometer is used, the solution volume can be expanded according to the actual need. Because the BCA detection results are dynamic, the absorption value has been increasing, spectrophotometer determination needs a long time, pay attention to the detection time is controlled within 10 minutes.

The carboxyl Fe3O4 NPs solution was diluted to 1mg/mL as a control. 1mL of each 4M@Fe3O4 and MG8@Fe3O4 were used for Measurement of sizer and then recycled.
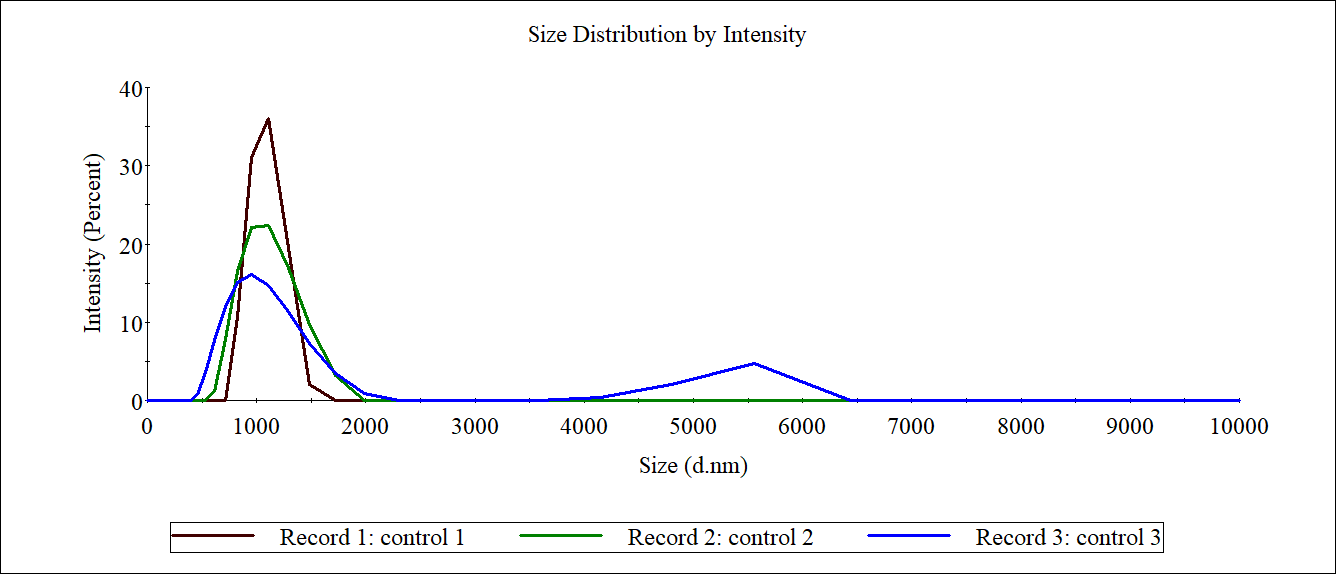
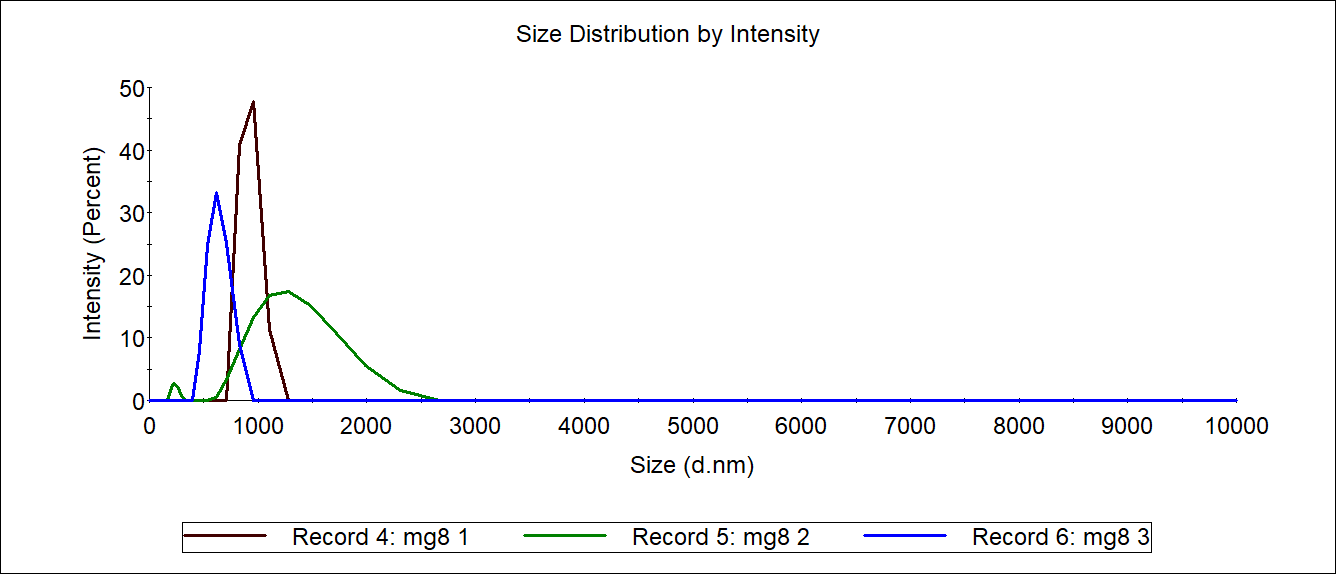

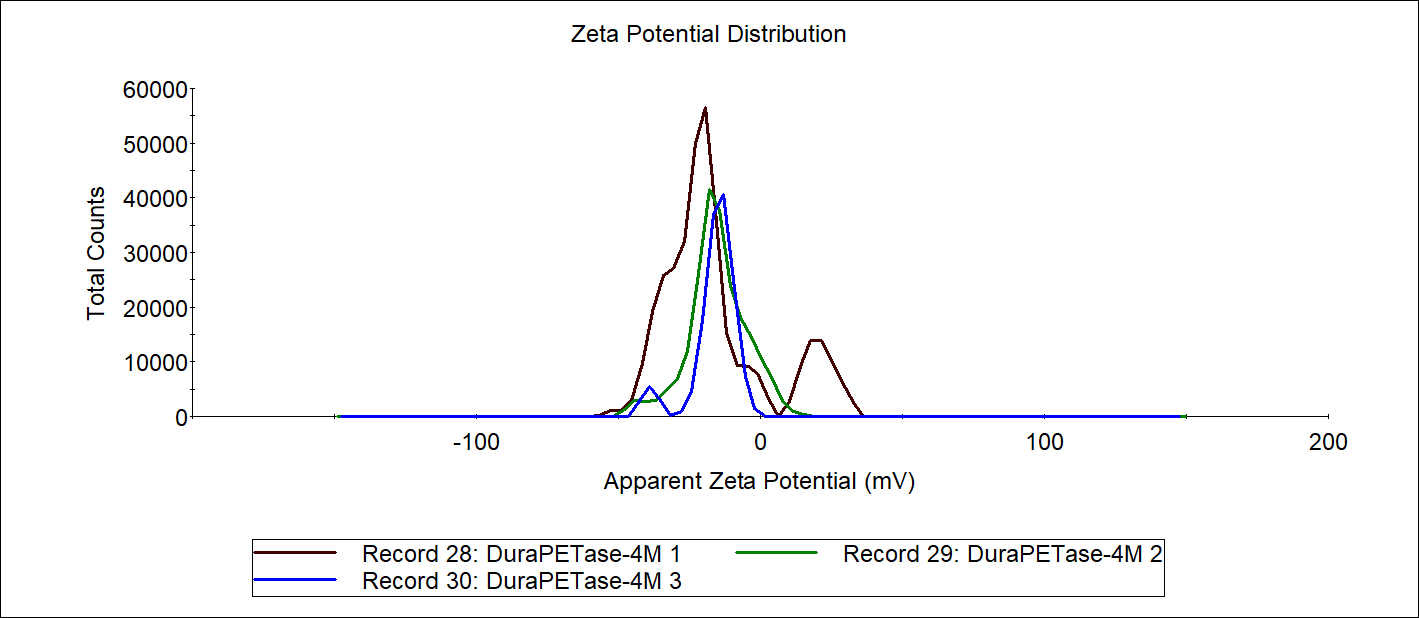
Adhesin-pET-28a plasmid was three competent cells Y01 C43 BLP.
After plate coating and overnight culture, three normal growths of the colony were selected from one plate for expansion culture to 8ml LB medium added 8μL kanamycin.
BLP and C43 were transferred into the medium for strain preservation, adding 0.5 volume of IPTG per thousand induction)37℃, 170rpm induction.
Glycerol bacteria Y01-4M and Y01-MG8 were inoculated with 500μl each into 200 mL medium (containing 200 μL kana of one thousandth antibiotic).

The induction result of Adhesin was determined by gel electrophoresis.
Y01-4M and Y01-MG8 were induced by adding 100 μL IPTG
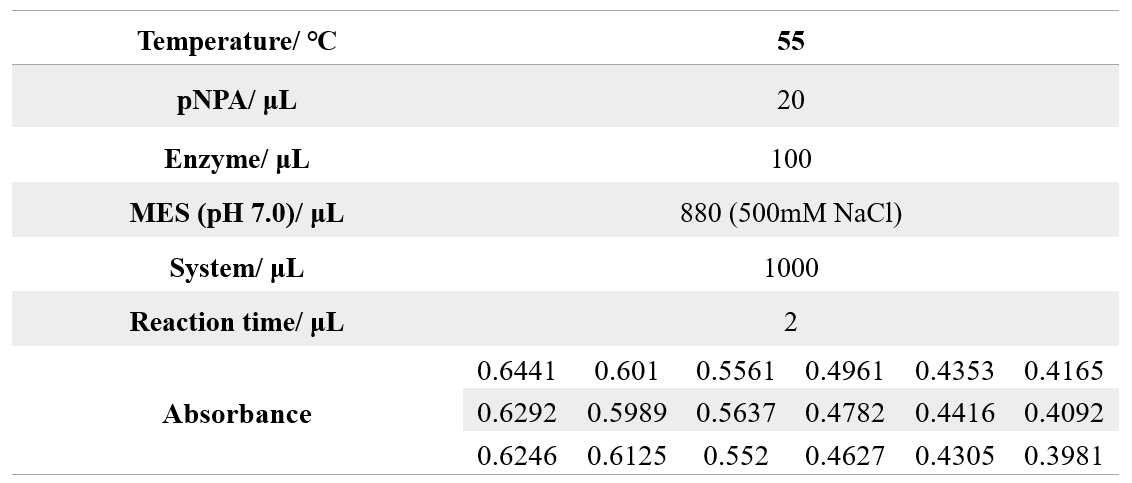
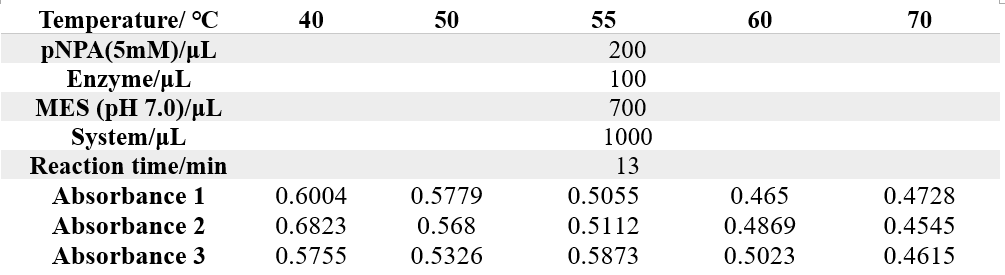
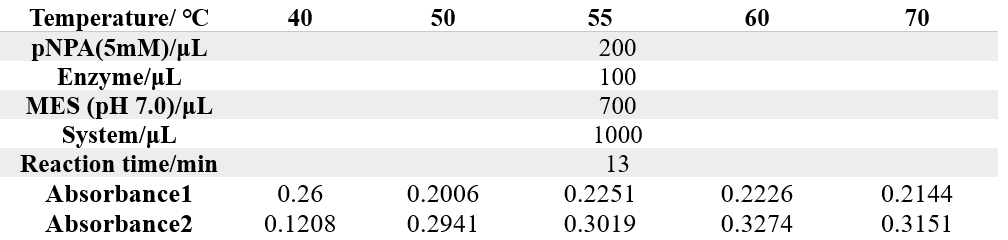
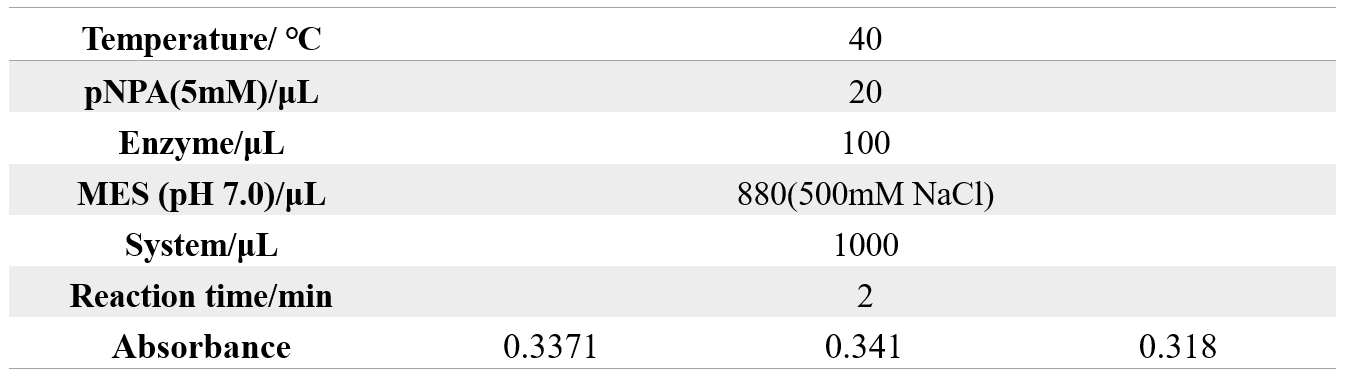
The reaction conditions of 4M are as follows:
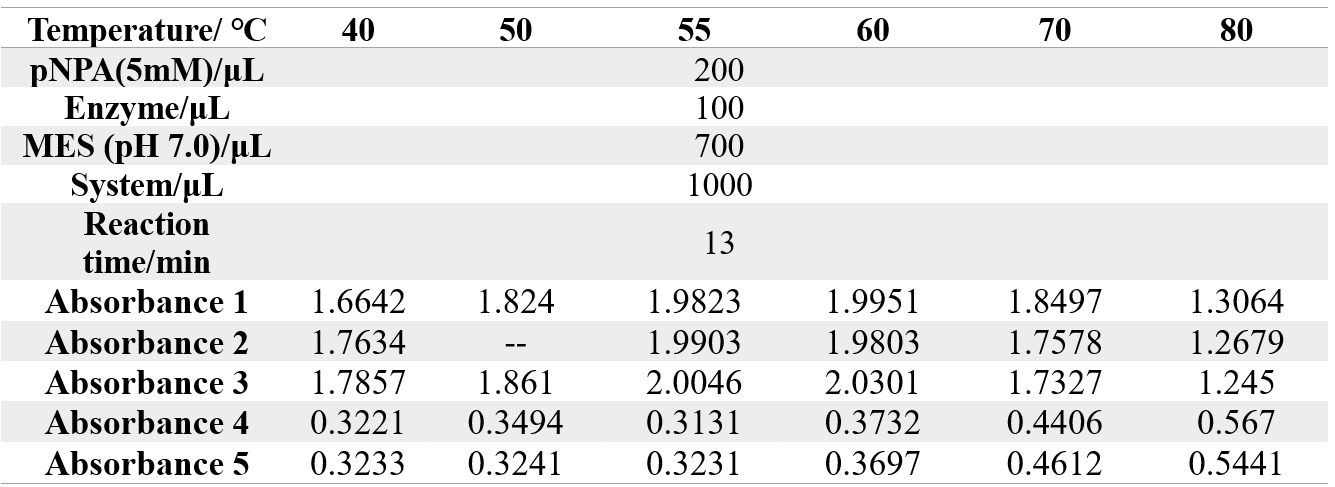
The reaction conditions of MG8 are as follows:
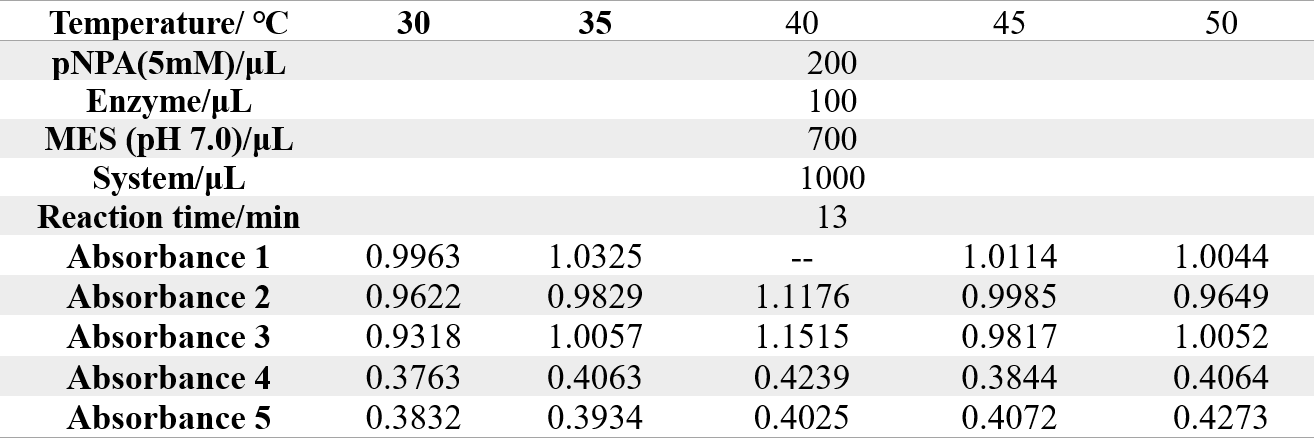
The reaction conditions of 4M are as follows:
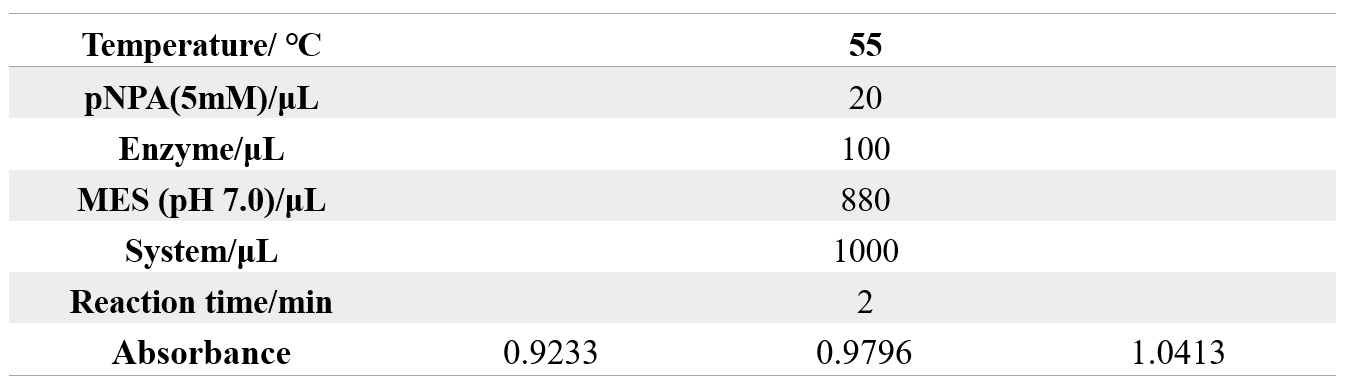
The reaction conditions of MG8 are as follows:
Inoculate 500 μL of glycerol Adhesins into 200ml of medium (containing 200 μL kana of one thousandth antibiotic).
The reaction conditions of 4M@Fe3O4NPs are as follows:

The reaction conditions of MG8@Fe3O4NPs are as follows:

The reaction conditions of 4M@Fe3O4NPs are as follows:
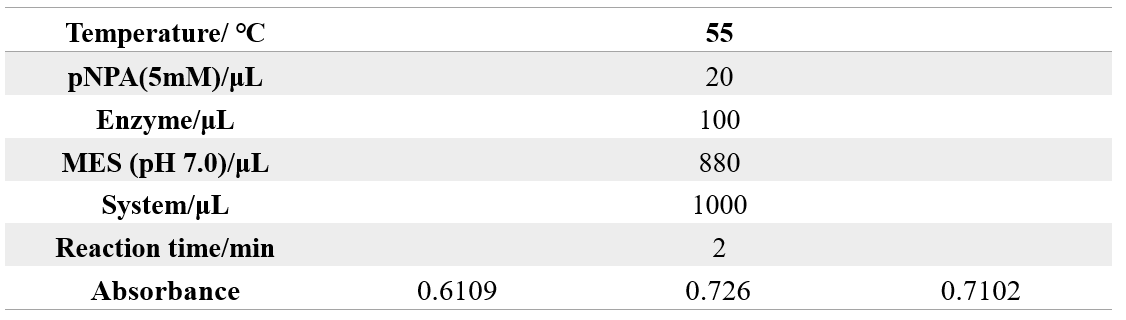
The reaction conditions of MG8@Fe3O4NPs are as follows:

The reaction conditions of 4M@Fe3O4NPs are as follows:

The reaction conditions of MG8@Fe3O4NPs are as follows: